By the Domainex Synthesis Group (Alicia Galván Álvarez, Claire Sear, Hugh Tawell, John May, Tenin Traore, Andrew Jones, Jonas Calleja and David Gibson)
In the latest edition of our blog series, we have focused on the following two recent publications, which describe highly useful synthetic transformations to achieve catalytic and enantioselective synthesis of stereogenic P(V) building blocks and a copper metallaphotoredox-mediated deoxytrifluoromethylation of alcohols:
- Enantioselective hydrogen-bond-donor catalysis to access diverse stereogenic-at-P(V) compounds. Katherine C. Forbes and Eric N. Jacobsen. Science, 2022, 376, 1230–1236.
- Deoxytrifluoromethylation of Alcohols. David W. C. MacMillan et al., J. Am. Chem. Soc. 2022, 144, 27, 11961-11968.
Catalytic and enantioselective synthesis of stereogenic phosphorus (V) centres
The facile stereoselective synthesis of molecules containing stereogenic phosphorus (V) centres is a long-lasting challenge in organic chemistry. Most of the existing methods rely almost entirely on the use of stoichiometric chiral auxiliaries to achieve diastereocontrol. Therefore, the development of asymmetric catalytic strategies is still a highly desirable goal. Some work has been recently reported towards this end, such as the catalytic nucleophilic reactions of chlorophosphoramidates (DiRocco et al.3) and phosphoramidites (Miller et al.4). However, these reactions are limited to a very specific class of nucleophiles that cannot be further displaced.
To overcome this, Jacobsen and co-workers1 envisioned that the catalytic and enantioselective installation of a nucleophile that could further serve as a leaving group would provide a general strategy for the synthesis of chiral P(V) targets. Dual H-bond donor catalysts have been successfully used in the literature to promote stereoselective nucleophilic substitution reactions via chloride-abstraction, and that was their catalyst of choice. The authors reported an enantioselective hydrogen-bond-donor catalysed synthesis of aryl chlorophosphonamidates that can be sequentially and stereospecifically displaced to obtain a broad range of stereogenic-at-P(V) molecules with a wide range of substitution patterns.
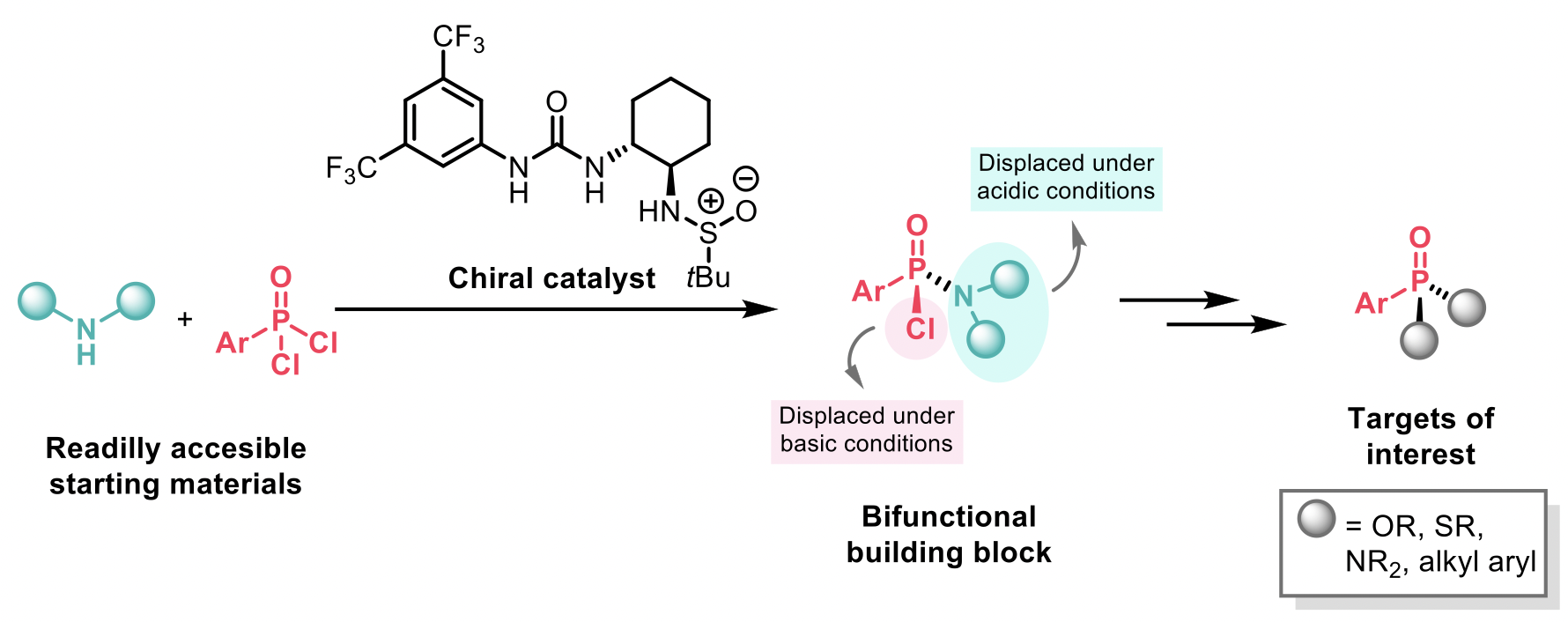
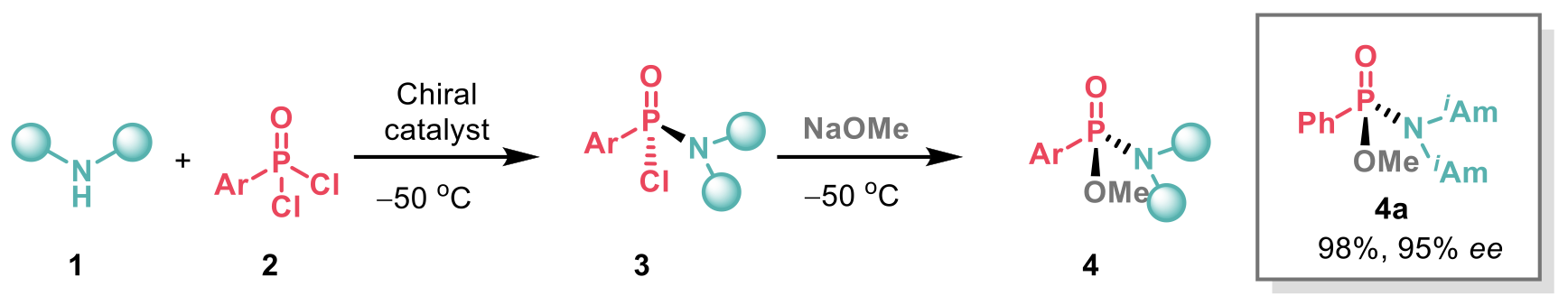
Investigation of the catalyst revealed that both the presence of the sulfinamide group and the H-bond donor were crucial in promoting high enantioselectivity. It was observed that chlorophosphonamidite intermediates 3 did not epimerised under the reaction conditions, but racemisation occurred slowly at room temperature over several hours. Therefore, the authors decided to quench 3 with NaOMe at low temperature to afford intermediates 4, which were found to have improved stability compared to 3. High levels of enantioselectivity were obtained by the use of diisoamylamine (see example 4a). Diisoamylamine was therefore selected to explore the scope of the reaction with different aryl phosphonic dichlorides 2. Alkyl phosphonic dichlorides, however, were not appropriate substrates for the transformation giving lower yields and poor ee.
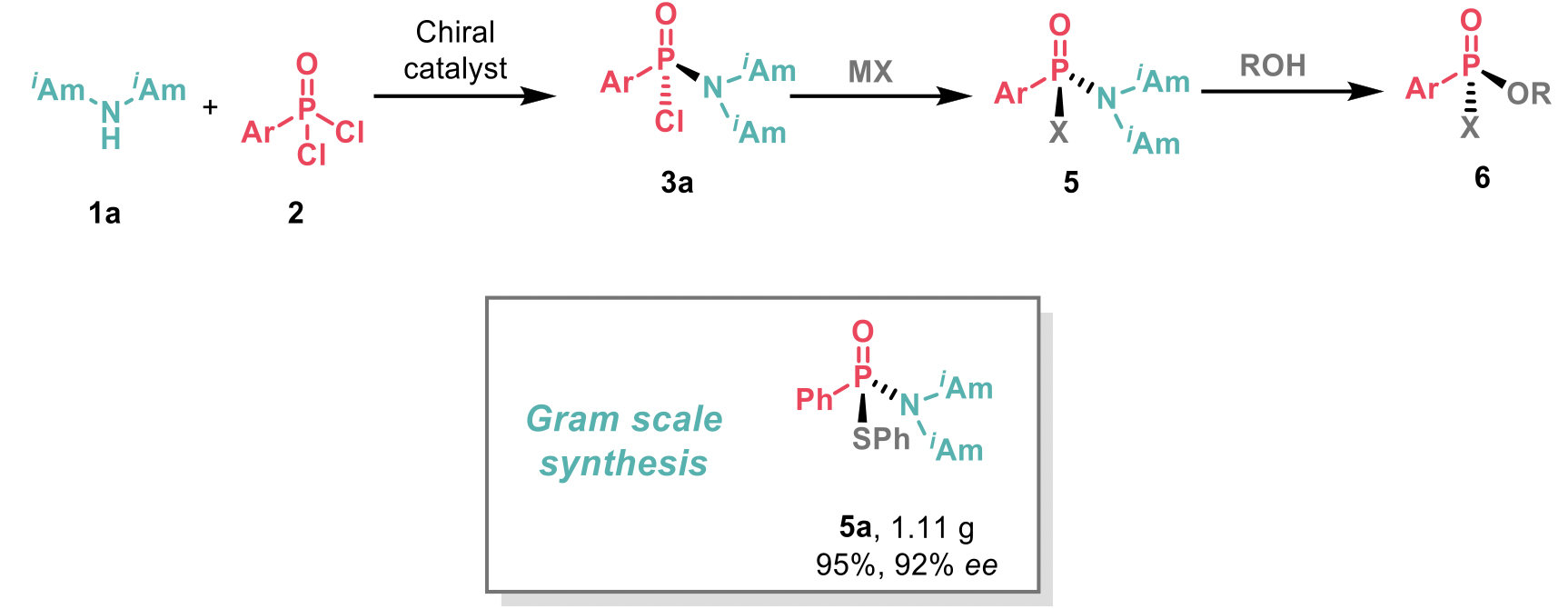
The authors also demonstrated the chloride displacement in chlorophosphonamidites 3 with different nucleophiles (MX) to achieve intermediates 5 with high levels of enantiospecificity. Reactions could be scaled up without any loss of enantioselectivity or yield, and 1.11 g of 5a was synthesised in one-pot. Intermediates 5 could also be further elaborated via an acid-mediated stereoinvertive displacement of the diisoamylamino group with alcohols (ROH), to obtain compounds 6.
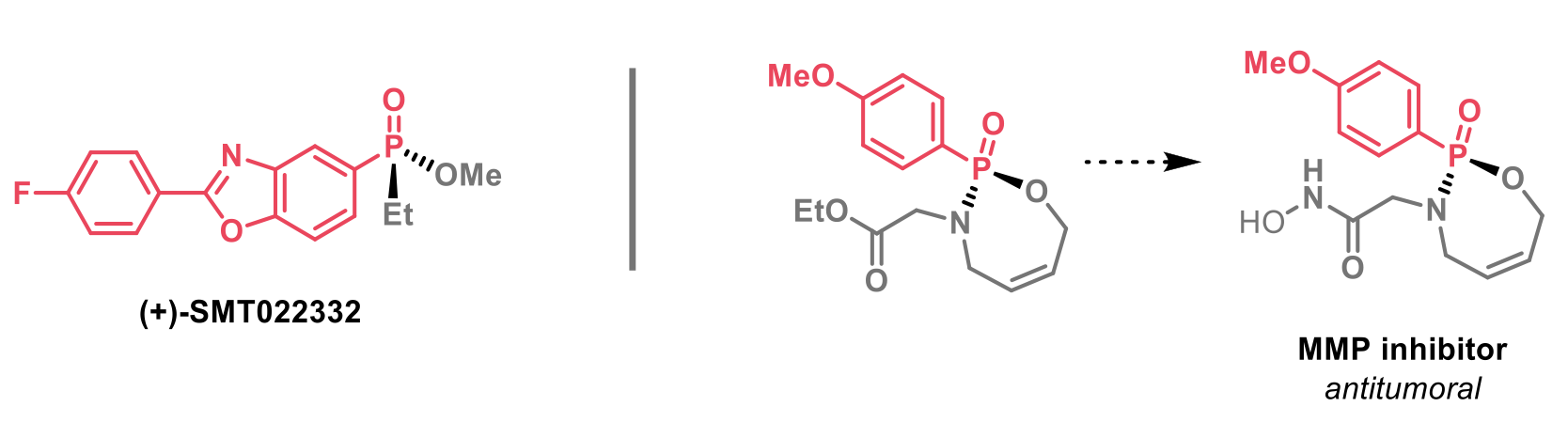
This methodology was successfully applied to the synthesis of (+)-SMT022332, a utrophion modulator developed as a potential treatment for Duchenne muscular dystrophy and also to the synthesis of a matrix metalloproteinase (MMP) inhibitor with demonstrated anticancer activity.
Copper metallaphotoredox-mediated deoxytrifluoromethylation of alcohols
Owing to the importance of fluorine in drug molecules due to its unique properties (i.e. ability to enhance pharmacokinetic properties), the necessity to develop robust chemical syntheses to access a wider range of fluorine containing molecules has never been more desirable. The trifluoromethyl (CF3) group has increasingly been incorporated in drug candidates due to the resulting increase in potency, and oral bioavailability, and decreased rate of oxidative clearance.
David W. C. MacMillan and his group2 have recently reported a general method for the deoxygenative trifluoromethylation of alcohols, wherein the alcohols are activated in situ by benzoxazolium salts. This results in the formation of an N-heterocyclic carbene (NHC)-alcohol adduct that enters the photoredox catalytic cycle to generate an alkyl radical which enters the copper catalytic cycle to generate the product.

The transformation conditions were robust and applicable to a range of substrates in yields ranging from moderate to good in most cases. The methodology was effective on simple primary, secondary and tertiary alcohols. In addition, the authors developed a strategy to install CF3 moieties to aromatic containing compounds by the use of an iterative functionalisation of diols. Initial arylation of the diol by conditions previously developed by the group formed the monoarylated intermediate that was successfully trifluoromethylated (see scheme below).
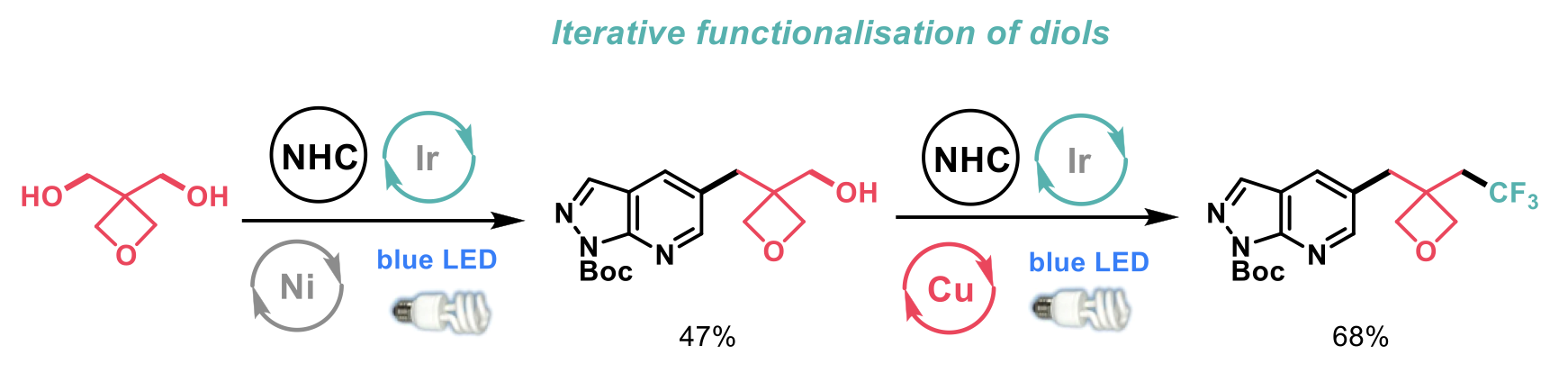
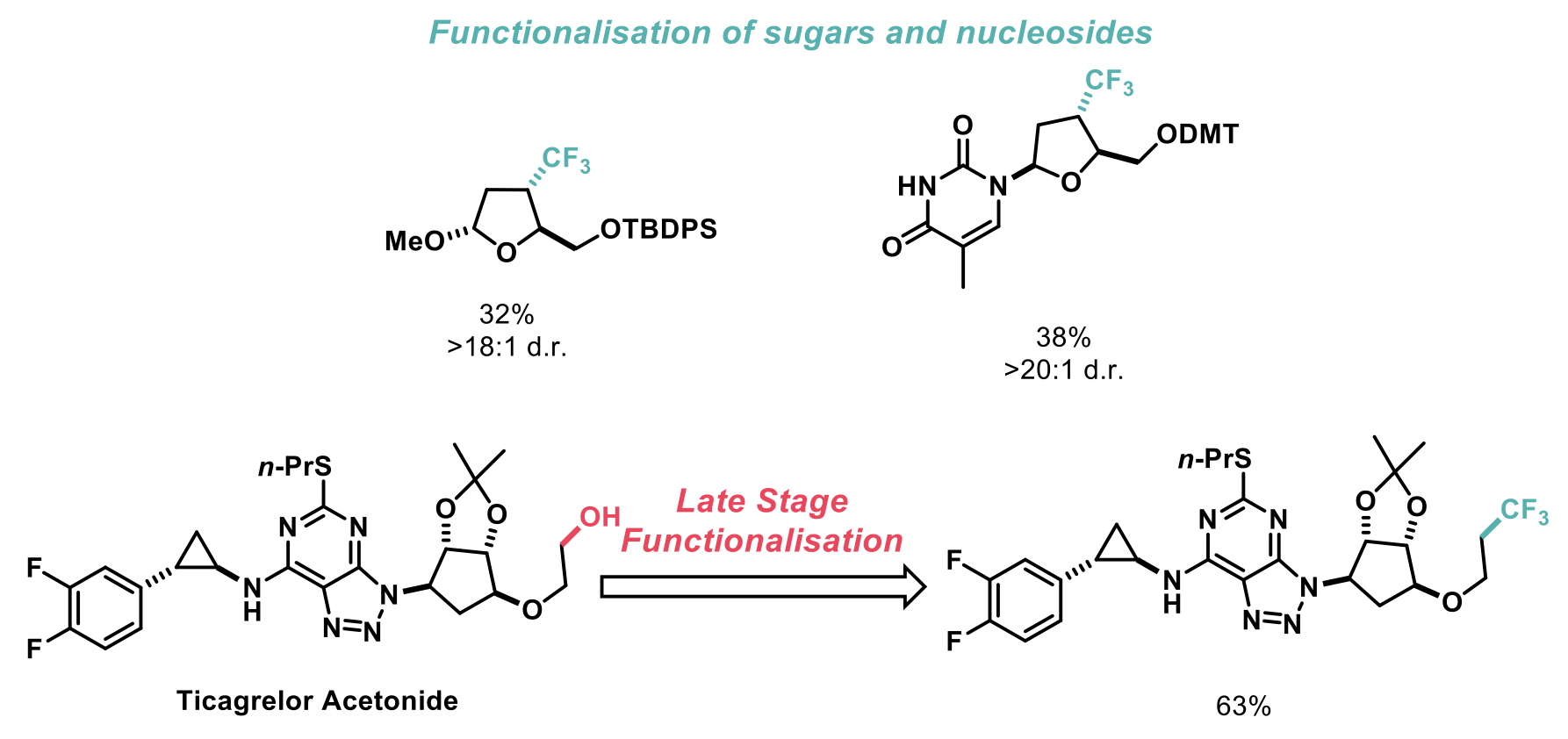
The authors also performed the trifluoromethylation of monosaccharides and the long-standing challenge of synthesising trifluoromethylated nucleoside derivatives. Finally, they tested the utility of the reaction in the context of late-stage functionalisation of pharmaceutical building blocks, and efficiently trifluoromethylated the cardiovascular drug Ticagrelor.
Additional references
3. D. A. DiRocco et al., Science 356, 426-460 (2017)
4. S. J. Miller et al., Science 371, 702-707 (2021)