- About
-
Services
-
Offerings
- ADME and Bioanalytical Sciences
- Analytical Chemistry
- Assay Development
- Biochemical Assays
- Biophysical Assays
- Cell Based Assays
- Computational Chemistry
- Fragment and Compound Screening
- Integrated Drug Discovery Services
- Medicinal Chemistry
- Protein Expression and Purification Services
- Structural Biology
- Synthetic Chemistry
- Virtual screening
- Research Phases
- Therapeutic Areas
- Target Classes
-
Approaches & Techniques
- CDH (Target Gene Fragmentation)
- Cryogenic Electron Microscopy (Cryo-EM)
- Differential Scanning Fluorimetry (DSF) and nanoDSF Services
- Direct-to-Biology (D2B)
- Flow Cytometry
- Fragment Based Drug Discovery (FBDD)
- FragmentBuilder
- Grating-Coupled Interferometry
- High Throughput Screening
- Isothermal Titration Calorimetry (ITC)
- LeadBuilder
- MicroScale Thermophoresis (MST) Services
- PoLiPa (Membrane Protein Solubilisation)
- Structure Based Drug Design (SBDD)
- Surface Plasmon Resonance (SPR)
- X-ray Crystallography
-
Offerings
- Library
- News & Events
- Careers
Computational Chemistry
Effective drug candidates by design

Computational chemistry expertise can improve significantly the efficiency of your drug discovery project by helping to design compounds that have a high probability of binding to your target and the right molecular and physical properties to ultimately demonstrate good bioavailability.
You can access our Computer-Assisted Drug Design (CADD) expertise on a stand-alone basis, or as part of a fully integrated project alongside our medicinal chemistry and biology teams. Our CADD experts use state-of-the-art software and proactively develop new insights that are tailored to the particular characteristics of the binding pocket of your disease target. We offer a wide range of services that can support your drug discovery project during all phases of pre-clinical research.
Hit Identification Phase
Our proprietary technology platforms allow us to find hits for you that are free of undesirable chemical functionality, and with highly developable molecular and physicochemical properties. Good-quality initial hits greatly improve the probability that a hit can be successfully and quickly optimised to a candidate drug molecule.
LeadBuilder
Domainex’s proprietary virtual screening platform, identifies potential chemical starting points for your drug discovery programme in a rapid and cost-effective manner. This platform can be used in protein structure-based or ligand-based modes. When protein structural data is not available for your target, it may be possible for Domainex to build a homology model for drug design purposes.
Visit our LeadBuilder page to find out more.
FragmentBuilder
Our unique, integrated Fragment-Based Drug Design platform, rapidly identifies high-quality fragment hits. Central to the success of this platform is the continuous evolution of our screening libraries. Our computational chemists ensure not only good coverage of chemical space but also optimisation of additional characteristics, such as shape, flexibility, three-dimensional character and pharmacophoric coverage. They also advise our chemists how to develop fragment hits into more potent compounds by designing compound arrays that explore potential protein-ligand interactions.
Visit our FragmentBuilder page to find out more.
Hit-to-Lead and Lead Optimisation Phases
At these stages in your project, our computational chemists will give you valuable insights into compound design, scaffold hopping, property profiling and the analysis of structure-activity relationships (SARs).
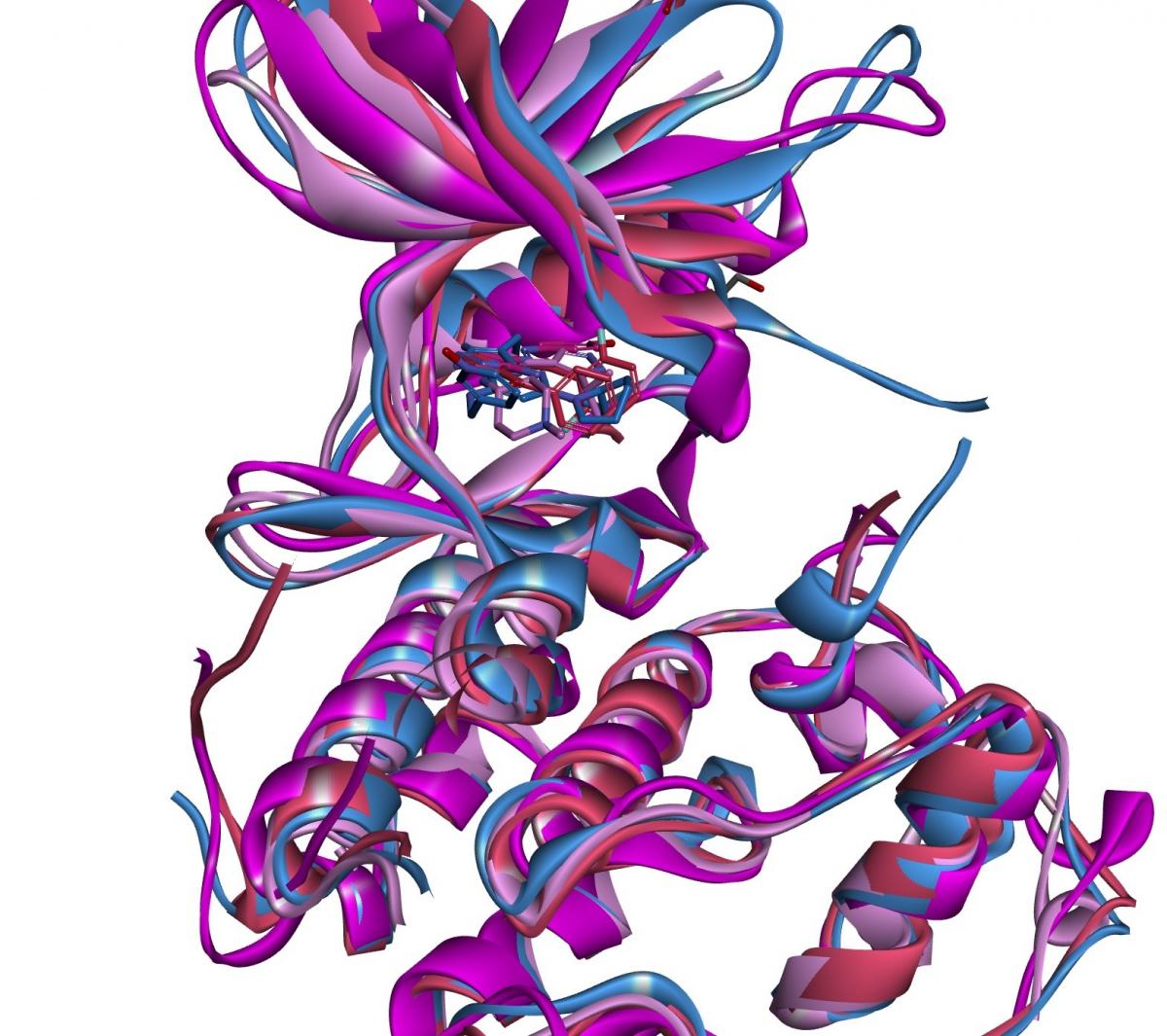
Structure-Based Drug Design (SBDD)
Our CADD team can analyse structural information about your nominated target, and design and help you prioritise ideas for prospective ligands. We can also overlay your target on related protein structures, using these comparisons to identify any differences which may enable the design of selective compounds.
Examples of projects where we have applied SBDD include:
- Der p 1: Protease inhibitors for the treatment of asthma
- Tankyrase: Inhibitors for the treatment of solid tumours and
- TBK1/IKKε: Kinases inhibitors for the treatment of interferonopathies
Ligand-Based Drug Design
Our CADD experts will work closely with you to analyse and interpret your data – for example, they could cluster your dataset and identify a subset of molecules for further development.
If you have a small dataset and want to further explore SARs around these chemotypes, our CADD team can search our curated proprietary databases of commercially available suppliers for nearest-neighbour analogues, which can be subsequently purchased and tested.
Property Profiling
Domainex also has several state-of-the-art property prediction tools, such as the ACDLabs software, that allows us to compare the predicted values for your compounds with those measured by our physical chemists. These comparisons can lead to improved predictive models for your series, and we can use these to devise structure-property relationships that are extremely informative in guiding drug design into the right property space.
Cresset aliance
![]()
Domainex can also offer additional computational chemistry services through its alliance with Cresset. Cresset offers a range of solutions including approaches centred on the application of the eXtended Electron Distribution (XED) force field to the design of new small molecule bioactive compounds. The XED approach improves traditional molecular mechanics by using a complex description of atoms to model charge away from atomic centres. This enables a more detailed description of electrostatics and excellent reproduction of intermolecular interactions.

Case studies
Start your next project with Domainex
Contact one of our experts today