By the Domainex Synthesis Group (Alicia Galván Álvarez, Claire Sear, Hugh Tawell, John May, Jonás Calleja Priede and Tenin Traore)
Domainex medicinal and synthetic chemists are always looking for reactions which can improve the efficiency of synthetic routes or allow access to novel targets. We were therefore pleased to see new examples of reactions that exhibit regio- and stereo- selectivity in the follow two recent papers and have focussed on these in this latest edition of our blog:
- Strain-Release-Driven Friedel–Crafts Spirocyclization of Azabicyclo[1.1.0]butanes. Jasper L. Tyler, Dr. Adam Noble, Prof. Varinder K. Aggarwal. Angewandte Chemie International Edition, 2022, 61, e202114235.
- Direct Synthesis of Enamides via Electrophilic Activation of Amides. Philipp Spieß, Martin Berger, Daniel Kaiser and Nuno Maulide. J. Am. Chem. Soc. 2021, 143, 28, 10524–10529
Strain-Release enabled Friedel-Crafts Spirocyclisation
Spiro N-heterocycles present beneficial physicochemical properties, structural novelty, three-dimensionality and enable the controlled spatial disposition of substituents. These properties have increased interest in these scaffolds from medicinal chemists working on drug discovery programs.
Aggarwal et al1 have recently published an article in ACIE where they report the strain-release-driven Friedel-Crafts spirocyclisation of azabicyclo[1.1.0]butane-tethered (ABB) (hetero)aryls towards the synthesis of azetidine spiro-tetralines. This acid-mediated reaction generates a complex dearomatised intermediate that can be converted to the Friedel-Crafts product or react as a Diels-Alder adduct with PTAD as dienophile in one-pot.
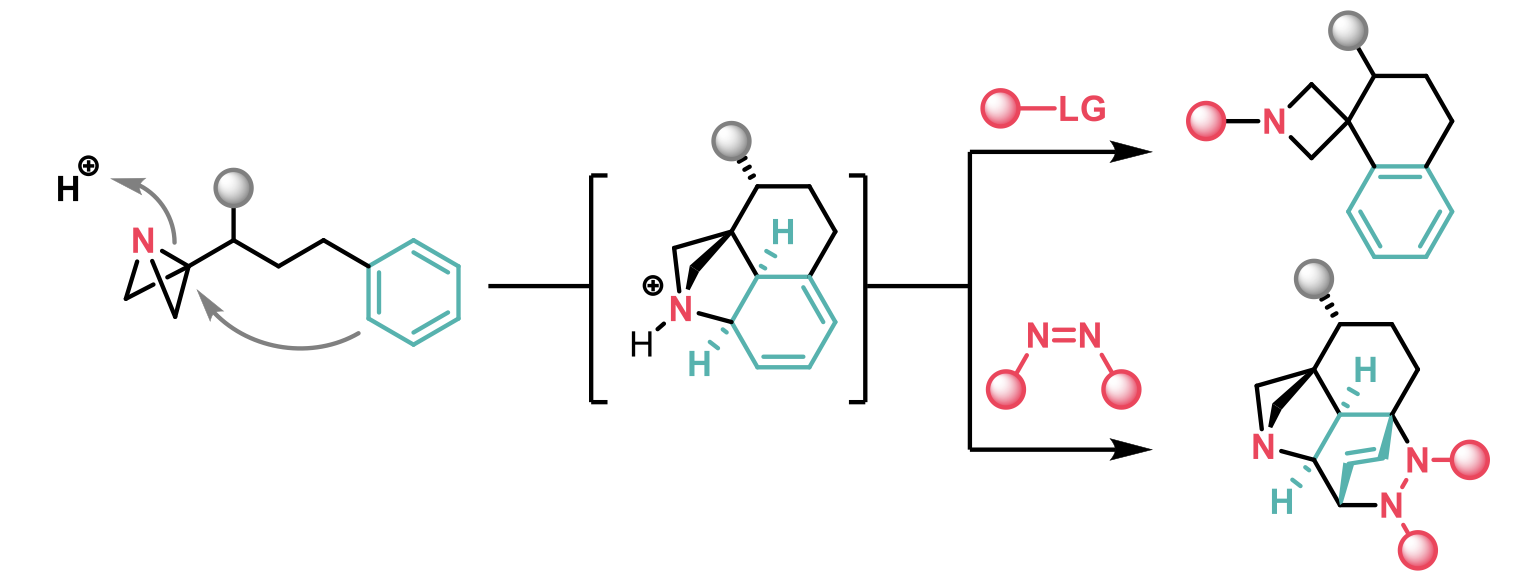
A broad range of ABB-tethered arenes were employed as well as trisubstituted alkenes, that enabled the spirocyclisation to be performed diastereoselectively. Complete regioselectivity was also observed in compounds containing a potentially reactive phenyl ring, and protected alcohols could be used, introducing a synthetic handle. The 2-naphthyl fragment was also a good nucleophile in the reaction and the spirocycle shown below, directly derived from the nonsteroidal anti-inflammatory drug nabumetone, could be synthesised in 46% yield.
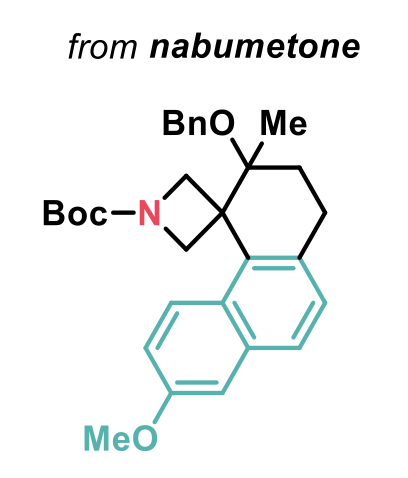
This method clearly highlights the potential of this strain release-driven spirocyclisation strategy in the synthesis of highly complex sp3-rich molecules.
From amides to enamides in one step
The exceptional nucleophilicity of enamines makes them a fundamental motif in the organic synthesis toolbox. However, due to their propensity to hydrolysis, enamides have recently experienced a revival as a less reactive but more stable surrogate of enamines. They are versatile reactants used in different fields such as transition-metal catalysis, photochemistry and asymmetric catalysis.
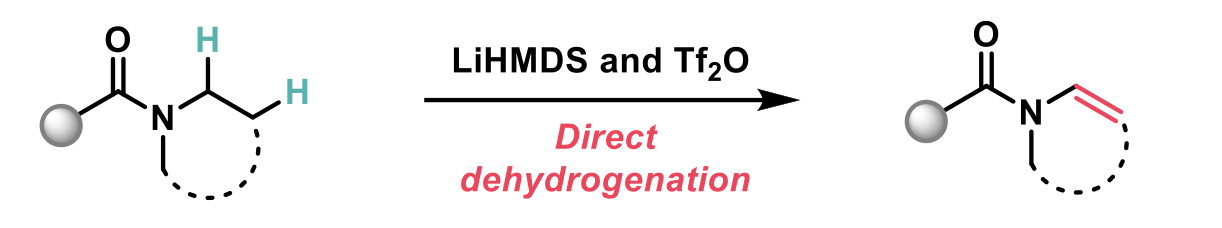
In this article recently published in JACS2, the authors describe a novel N-dehydrogenation of amides to achieve enamides, that does not require prefunctionalisation. This reaction proceeds through the electrophilic activation of the amide and utilises the unusual combination of LiHMDS and triflic anhydride, which serves as both the electrophilic activator and the oxidant.
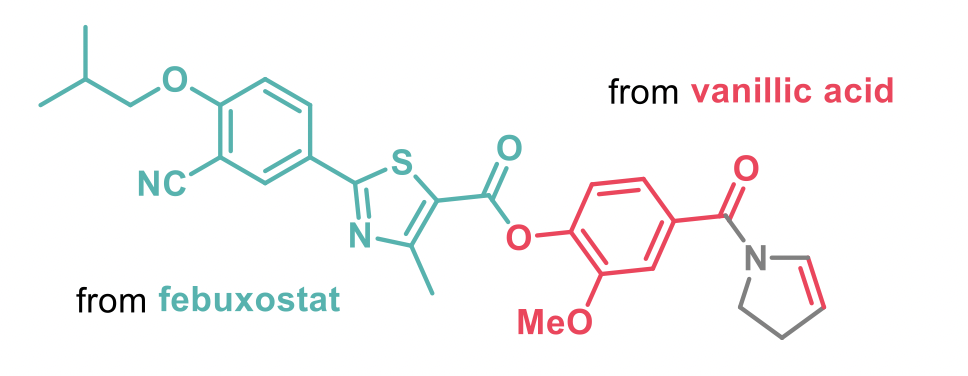
The reaction described is chemo- and regioselective with a broad substrate scope and very operationally simple. Even a functional-group heavy conjugate of vallinic acid and febuxostat provided the desired dehydrogenated product.
Further functionalisation reactions were performed on the enamides generated, including cycloaddition reactions, ring deconstruction or ring functionalisation to demonstrate the utility of the products formed.
Mechanistic studies were performed which suggested that the mechanism involved activation of the amide to an iminium triflate acidifing the N-α-hydrogen, after which a deprotonation step takes place, leading to extrusion of the triflinate anion and subsequent elimination to produce the observed enamide products.
This novel reaction allows access to enamides enabling the synthesis of highly complex organic molecules.
We hope you found this interesting! Look out for our next synthesis review which will be available on our website soon, and get in touch to find out how we can employ the latest synthetic approaches to solve your drug discovery challenges.