- About
-
Solutions
-
Services
-
Biosciences
- PureDisc™ Enabled Membrane Protein Production
- Assay Development and Transfer
- Biochemistry / Enzymology
- Biomarker Services
- Biophysics
- Cell-Based Assays
- Compound screening and cascade design
- Cryo-EM
- Fibrosis Assays
- Project Management and Consultancy Services
- Protein Expression
- Structural Biology
- X-ray Crystallography
- Chemistry
- Integrated Drug Discovery
- Computer Aided Drug Design
- Hit ID and Screening Services
- DMPK Services
-
Biosciences
- Target Classes and Modalities
- Therapeutic Areas
-
A-Z
- A
- B
- C
- D
- E
- F
- G
- H
- I
- K
- L
- M
- N
- O
- P
- R
- S
- T
- V
- X
-
Services
- Library
- News & Events
- Careers
- PureDisc™
X-ray Crystallography
Protein structure generation and analysis
X-ray crystallography is the gold standard technique for protein/protein-ligand structure determination, providing detailed information at high resolution. Domainex can couple extensive structural biology expertise with the in-house facilities to undertake high-throughput screening of thousands of crystallisation conditions to generate apo or holo-structures for both small and large molecule drug discovery programmes.
Domainex offers macromolecular X-ray crystallography services in support of drug discovery efforts.
- Gene to structure structural support with mid-way entry points
- High quality protein expression
- Structure elucidation of biological drug molecules
- Soaking systems and co-crystallization
- Hundreds-thousands of crystallisation conditions tested using in-house liquid handling and robotics
- Crystal imaging for efficient workflows
- Structural biology of ternary complexes for bivalents and molecular glues
- Close collaboration with medicinal and computational chemistry for structure-based drug design
- Structural support for fragment-based drug discovery and crystallographic Fragment screening
- Regular industrial access to synchrotron sources in the UK and throughout Europe
X-ray crystal structures of protein targets in complex with small molecules for structure-based drug discovery (SBDD) and SAR-guided design
X-ray crystal structures giving detailed insight into how small molecules interact with the target protein can be game-changing for drug discovery projects, enhancing what can be achieved using molecular modelling with experimental data, and providing detailed information for medicinal chemistry design. Domainex scientists have > 20 years of experience in structure determination of proteins in complex with small molecules. Combining our in-house protein production expertise with high throughput crystallisation and regular access to state of the art synchrotron sources in the UK and throughout Europe, we regularly obtain high resolution structures (2.5 Å or better) and work closely with medicinal and computational chemistry to guide drug design for step-changes in potency, selectivity or improving ADME properties. Our structural biologists work both in the early stages with hit finding for crystallographic fragment screening, structural biology for fragment or small molecule hits as well as more advanced medicinal chemistry programmes.
X-ray crystal structures of biological drug molecules
X-ray crystallography is well suited to providing critical data during development of biological drugs.
Monoclonal antibodies, single-chain variable fragments (scFv), nanobodies (from camelids) and variable novel antigen receptors (VNAR from sharks), to name but a few, have all become commonly used therapeutic, diagnostic and research tools.
Our protein sciences team here at Domainex can express and purify these complex biological molecules, along with interacting partners such as receptor ectodomains, and our biophysics team can provide KDs to define binding affinities.
Here at Domainex, we have experience in providing high resolution X-ray crystal structures of such biological molecules in complex with their targets. Protein complexes are formed and then purified using size exclusion chromatography (SEC), prior to high throughput crystallisation screening to find the optimal precipitant and incubation conditions for growing high quality protein crystals. Once building and refinement of the atomic coordinates are complete, we can provide detailed analyses of residues forming the epitope and/or paratope, including a full description of their interactions.
Proteins such as insulin which is used to treat diabetes and thrombin which is used to reduce bleeding during surgery are examples of approved biological drugs. Here at Domainex we have experience in using X-ray crystallography to assist our clients in the approval process, for example, by providing high resolution crystal structures of natural and recombinant variants of a chosen target protein. We then undertake a detailed comparison of the resulting X-ray structures, providing in depth reports for direct submission to the relevant regulatory body.
X-ray crystal structures of PROTAC and molecular glue-driven ternary complexes
X-ray crystallography has played a key role in recent years in the development and understanding of degrader drugs such as PROTACS and molecular glues. Published examples include cereblon (CRBN) in complex with mezigdomide and IKZF1 ZF2 (8RQC; https://doi.org/10.1038/s41467-024-52871-9) and the EloBC-VHL-CDO1 complex bound a molecular glue (8VL9; to be published).
Here at Domainex our protein sciences team have the expertise to express and purify your protein of interest (POI) and E3 ligase. We have E3 ligases such as CRBN and von Hippel–Lindau protein (VHL) available to buy immediately. Ternary complexes are formed and their stability is demonstrated using techniques such as size exclusion chromatography (SEC) and mass photometry.
Crystal soaking systems
Crystal soaking systems are utilised for high throughput ligand screening and at Domainex we aim to develop these systems early in each programme. Ideally, a reproducible single crystal at a resolution greater than 2.5 Å is desirable for a successful soaking system. The advantages of soaking systems are that several crystals from one drop can be utilised in several different soaking experiments with various ligands. Soaking systems can be applied to both fragments and more elaborate compounds.
At Domainex, we have developed successful soaking systems for multiple projects which have enhanced the structure activity relationships (SAR) of chemical series and support drug design. A recent integrated programme generated over 30 target protein structures with resolution better than 1.8 Å which enabled compound optimisation to identify a selective lead candidate.

Co-crystallisation
What happens if you can’t develop a soaking system and a target only co-crystallises? At Domainex, we also regularly develop co-crystallisation optimisation screens for high-throughput screening. These screens are generally fine-tuned as a grid screen around the original crystallisation conditions with optimisation of cryoprotectant and crystal freezing methods for each system. For example, during a fragment-based drug discovery project targeting the methyltransferase, G9a, we generated four co-crystal structures which enabled our scientists to design compounds with a 10-fold improvement in binding affinity after just one round of fragment elaboration.
We often find that both soaking and co-crystallisation strategies work well together.


Image kindly provided by the Diamond Light Source
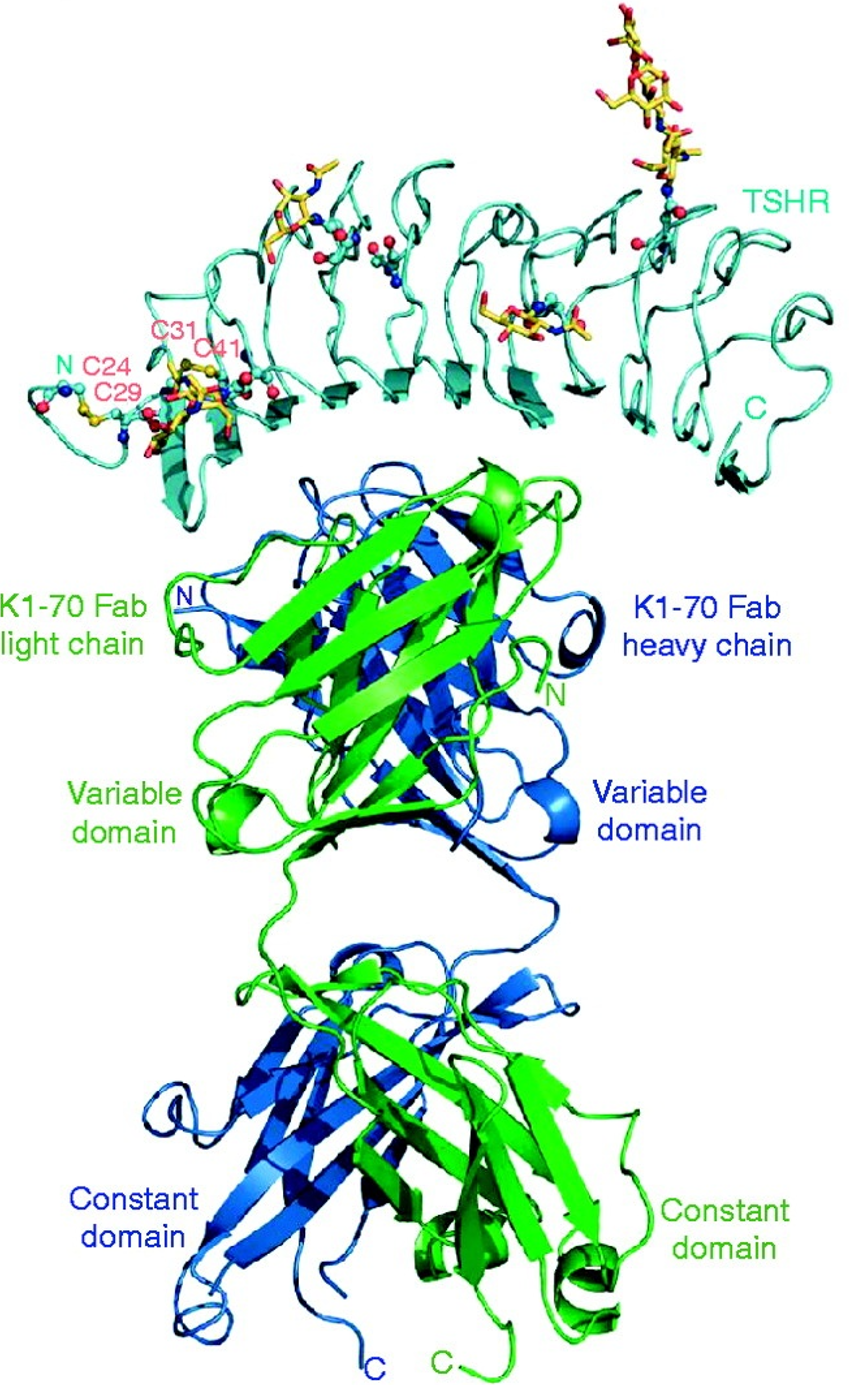
Example published structure: The complex of the TSH receptor leucine-rich repeat domain (TSHR LRD) and the human monoclonal autoantibody K1-70 Fab at 1.9 Å resolution (Journal of Molecular Endocrinology (2011) 46, 81–99)
Case studies
Start your next project with Domainex
Contact one of our experts today