- About
-
Solutions
-
Services
-
Biosciences
- PureDisc™ Enabled Membrane Protein Production
- Assay Development and Transfer
- Biochemistry / Enzymology
- Biomarker Services
- Biophysics
- Cellular Biology
- Compound screening and cascade design
- Cryo-EM
- Fibrosis Assays
- Project Management and Consultancy Services
- Protein Expression
- Structural Biology
- X-ray Crystallography
- Chemistry
- Integrated Drug Discovery
- Computer Aided Drug Design
- Hit ID and Screening Services
- DMPK Services
-
Biosciences
- Target Classes and Modalities
- Therapeutic Areas
-
A-Z
- A
- B
- C
- D
- E
- F
- G
- H
- I
- K
- L
- M
- N
- O
- P
- R
- S
- T
- V
- X
-
Services
- Library
- News & Events
- Careers
- PureDisc™
Computational Chemistry Services
Computer-aided drug design for effective drug candidates

Computational chemistry is a powerful accelerator for modern in silico drug discovery.
At Domainex, our Computer-aided drug design (CADD) team combines state-of-the-art cheminformatics software with deep scientific expertise to design compounds with a high probability of binding to your target and with the right molecular properties for successful development. Our CADD services are available on a stand-alone basis or as part of a fully integrated drug discovery programme with our medicinal chemistry and biology teams.
Our Capabilities
Cheminformatics & Data Analysis
Turning complex datasets into clear insights through the integration of advanced cheminformatics software.
- Automated data retrieval pipeline
- Cluster and visualise chemical space
- Interpret structure activity relationship (SAR) trends to identify key design drivers
- R-group analysis
- Prioritise compounds with the greatest potential
- Identify nearest-neighbour analogues from curated supplier databases for rapid experimental follow-up
Artificial Intelligence & Machine Learning (AI/ML)
Accelerating discovery with predictive modelling services that incorporate the latest advances in AI and Machine learning, guided by Domainex expertise.
- Build custom AI/ML models tailored to your project
- Predict activity, selectivity, and absorption, distribution, metabolism, excretion and toxicity (ADMET) properties
- Identify non-obvious SAR
- Support data-driven decision making at every stage of discovery
Ligand-Based Drug Design
When structural data is limited, ligand-based drug design (LBDD) allows you to build predictive models and design promising scaffolds.
- Build predictive models from your ligand datasets
- Identify and optimise promising scaffolds
- Expand SAR exploration with focused compound sets
- Pharmacophore/Shape/Electrostatic Modelling
- Scaffold Hopping
- Leverage proprietary and commercial chemical databases for rapid design cycles
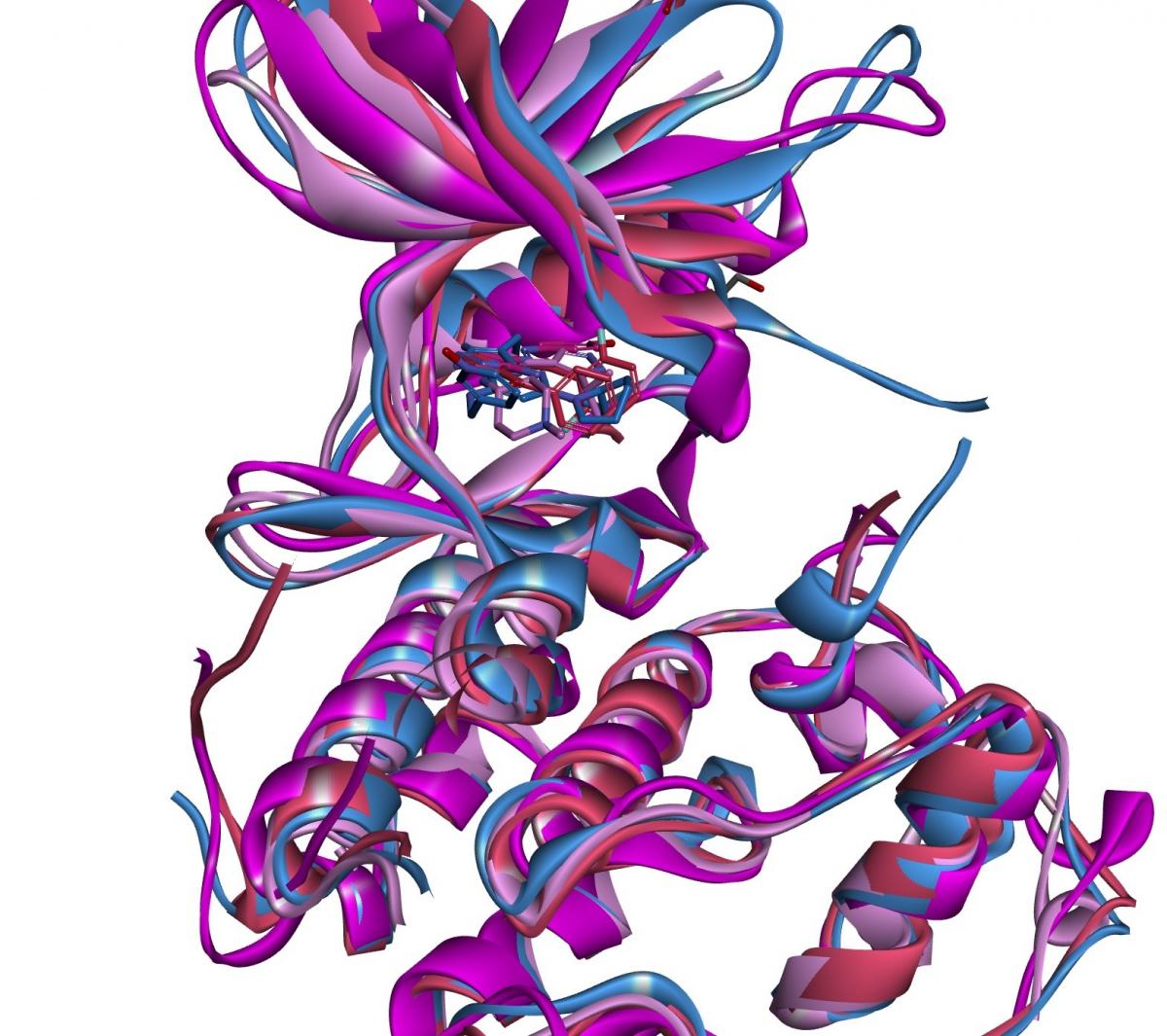
Structure-Based Drug Design
Exploit protein structures for precision design using Domainex’s structure-based drug design (SBDD) services.
- Molecular docking, pharmacophore modelling, and dynamics simulations
- Torsional analysis to identify strain in bound ligand conformations
- Binding site detection and druggability assessments – in combination with Molecular Dynamics (MD) enables the identification of allosteric/cryptic binding sites
- AI-enabled protein–ligand co-folding and traditional homology modelling when crystal structures are not available
- Cross-target structural comparisons to design selective ligands
- Water mapping to identify key hydration sites driving affinity and selectivity
- Accurate physics-based assessment of binding affinities via Free Energy Perturbation (FEP) and Fragment Molecular Orbital (FMO) Analysis
- AI-guided blind docking to discover novel binding pockets without prior assumptions
- Machine Learning (ML) approaches to identify potentially reactive cysteines
- Applied to diverse targets including:
ADMET Modelling & Property Prediction
Guiding compounds into the right property space, through an in depth understanding of Absorption, Distribution, Metabolism, Excretion and Toxicity.
- Prediction of efflux, Kp,uu, permeability, pKa and logD
- AI/ML enabled modelling of rates and sites of metabolism (CYP450 isoforms and hAOX)
- hERG, AMES and photoxicity modelling
- Early flagging of undesirable features
- Integration of computational predictions with experimental measurements

Technology Platforms
LeadBuilder™
Proprietary virtual screening platform for rapid, cost-effective identification of chemical starting points. Supports both structure-based and ligand-based modes, with homology modelling available when no structure is present.
FragmentBuilder™
Our integrated fragment-based drug design (FBDD)platform, featuring continuously evolving fragment libraries optimised for shape, flexibility, 3D character, and pharmacophoric coverage. Designed to generate fragment hits that can be efficiently grown into leads.
Software & Tools
Software & Tools
Domainex computer-aided drug design team have access to industry-leading platforms, including:
- Schrödinger – advanced molecular modelling, docking, and predictive design
- Pharmacelera – innovative ligand-based design using high-dimensional molecular descriptors


Why Work With Domainex?
- Integrated expertise – seamless collaboration between assay biology, medicinal chemistry, and CADD teams
- Proprietary platforms – LeadBuilder™ and FragmentBuilder™ accelerate discovery
- Best-in-class software – access to Schrödinger and Pharmacelera
- Proven success – delivered across diverse targets including enzymes, kinases, and protein–protein interactions
Case studies
Start your next project with Domainex
Contact one of our experts today