- About
-
Solutions
-
Services
-
Biosciences
- PureDisc™ Enabled Membrane Protein Production
- Assay Development and Transfer
- Biochemistry / Enzymology
- Biomarker Services
- Biophysics
- Cellular Biology
- Compound screening and cascade design
- Cryo-EM
- Fibrosis Assays
- Project Management and Consultancy Services
- Protein Expression
- Structural Biology
- X-ray Crystallography
- Chemistry
- Integrated Drug Discovery
- Computer Aided Drug Design
- Hit ID and Screening Services
- DMPK Services
-
Biosciences
- Target Classes and Modalities
- Therapeutic Areas
-
A-Z
- A
- B
- C
- D
- E
- F
- G
- H
- I
- K
- L
- M
- N
- O
- P
- R
- S
- T
- V
- X
-
Services
- Library
- News & Events
- Careers
- PureDisc™
Der p 1: Protease Inhibitors for the treatment of asthma
An inhaled treatment for allergic asthma
Domainex helped develop a new inhaled treatment for allergic asthma targeting Der p 1
![]()
Challenge
It has been estimated that more than 339 million people suffer from asthma1 and, according to WHO estimates, there were 417,918 deaths due to asthma globally in 2016.2 Established asthma drugs (β2 agonist bronchodilators, corticosteroids and anti-leucotrienes) treat the symptoms but do not address the causes of asthma. Therefore, a drug that can treat the triggers of asthma by blocking the bioactivity of allergens would be highly desirable.
Proteins produced by house dust mites (HDM) are one of the commonest causes of domestic allergy and are a major trigger for asthma attacks. The so-called 'Group 1 allergens' produced by HDM are globally prevalent and clinically important triggers of allergic asthma. Group 1 HDM allergens are excreted cysteine peptidases whose proteolytic activity triggers essential steps in the allergy cascade, and they are believed to be the proteins that initiate the allergic response, not just to other HDM allergens, but to a wider range of allergic insults.
Medicinal chemists at Domainex helped to develop inhibitors of the catalytic activity of these proteases (focusing on Der p 1) with properties suitable for inhaled delivery.
Solution
Prior to the commencement of the programme, the only reported inhibitors of Der p 1 were irreversible peptide based acyloxymethyl ketone inhibitors such as 1 (Figure 1).3 Peptides are generally poor oral drugs due to the metabolic instability of the peptide bonds, high molecular weight and polar surface area, large number of hydrogen bond donors and acceptors and rotatable bonds. Conversely, they are ideal for inhaled drugs since their properties mean they are retained well in the lung, increasing the duration of action, and they have limited systemic exposure (due to poor absorption and high metabolism). However, given that asthma treatment requires chronic drug administration, we considered that compounds having a reversible mechanism of action would be more desirable and developable since irreversible inhibitors may cause idiosyncratic toxicity. Therefore, we initially worked on modifying the cysteine binding motif which led to the reversible amino-ketone 2. However, we were unable to improve the potency of this amino-ketone series, so we investigated alternative cysteine binding motifs and this ultimately led to the identification of a potent pyruvamide lead compound (compound 4).
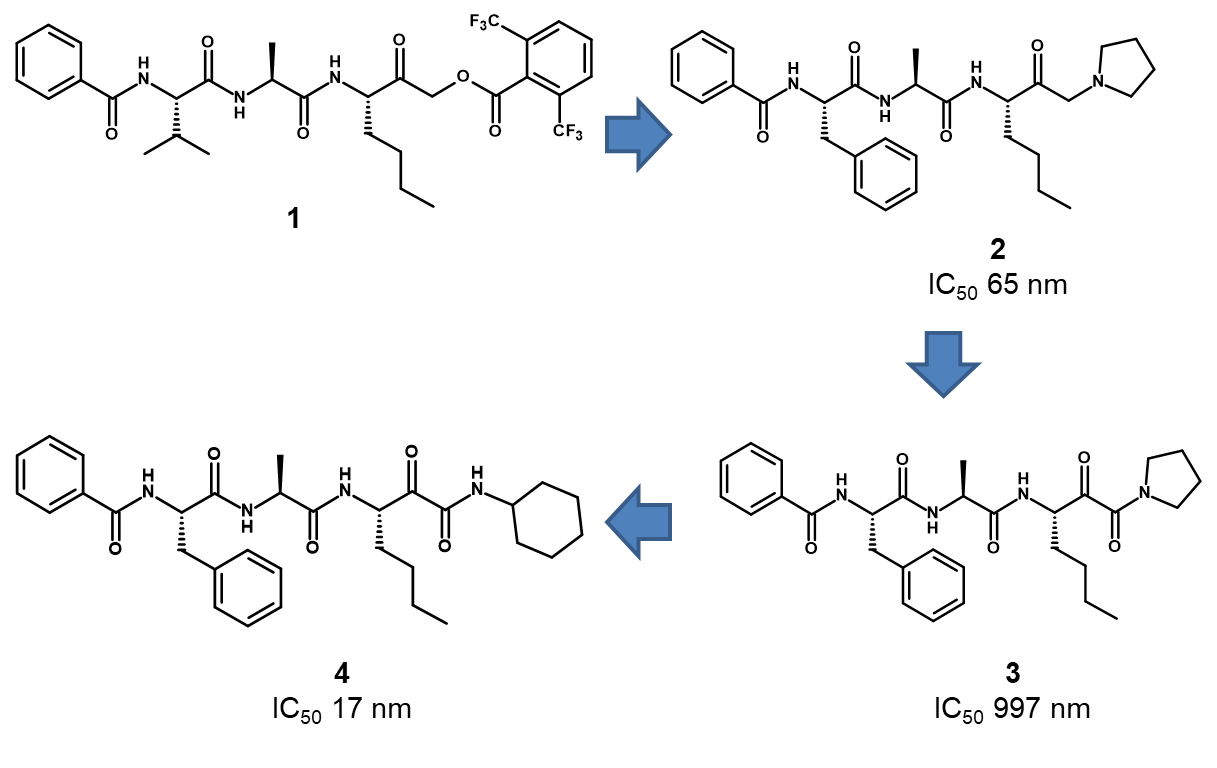
Compound 4 had the required potency and many of the properties required for an inhaled drug. However, it lacked selectivity over the closely related human cysteine peptidase, cathepsin B (IC50 16nM). Additionally, we found that compound 4 was extensively (>50%) degraded by airway macrophages over a 2 hour period, suggesting that its duration of action in vivo would not be sufficient.
In order to design compounds with improved selectivity, we generated a model of 4 bound to Der p1 (Figure 2) based on a crystal structure of Der p 1 in the public domain (pdb code: 2AS8). The electrophilic carbonyl of the pyruvamide was positioned to form an interaction with the catalytic cysteine residue (Cys 34) and the minimised structure formed a number of putative hydrogen bonds (see Figure 3).
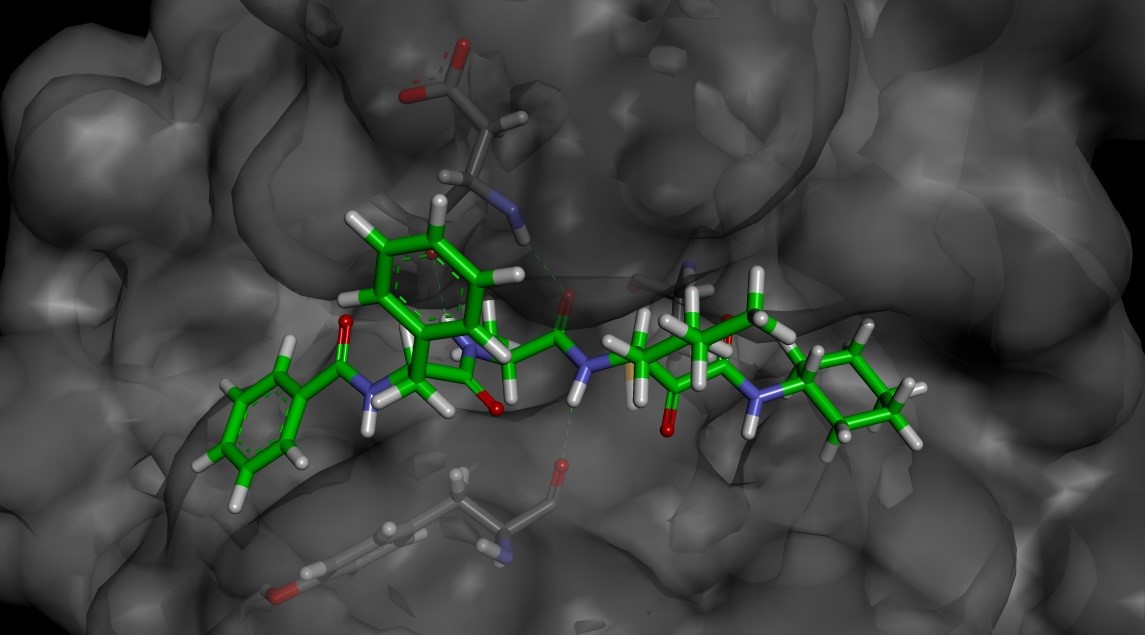
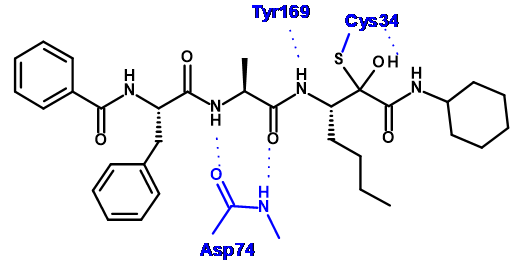
The P’ cyclohexyl group could be replaced with a benzyl group without a significant impact on the potency or selectivity. Therefore, these groups were both used for comparison when assessing changes to P1, P2 and P3 (Table 1). Replacing the P1 n-butyl group with a bulkier i-Pr had little effect on potency or selectivity, but did improve the stability to airway macrophages (~30% degraded over a 2 hour period). However, increasing the bulk further to a t-butyl group (compound 7) led to a decrease in potency.
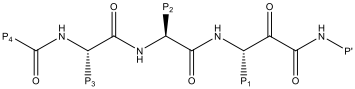
| Compound | P1 | P2 | P3 | P4 | P’ | Der p 1 IC50 (nM) | Cat B IC50 (nM) | LogD7.4 |
|---|---|---|---|---|---|---|---|---|
| 4 | n-Bu | Me | benzyl | Ph | cyclohexyl | 8 | 17 | |
| 5 | i-Pr | Me | benzyl | Ph | cyclohexyl | 18 | 52 | |
| 6 | i-Pr | Me | benzyl | Ph | benzyl | 12 | 50 | |
| 7 | t-bu | Me | benzyl | Ph | cyclohexyl | 9167 | Not determined | |
| 8 | i-Pr | Me | t-butyl | Ph | cyclohexyl | 14 | 378 | 3.9 |
| 9 | i-Pr | Me | C(Me)2Ph | Ph | benzyl | 42 | 446 | |
| 10 | i-Pr | n-Pr | benzyl | Ph | cyclohexyl | 164 | 67 | |
| 11 | i-Pr | Me | t-butyl | 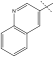 |
benzyl | 18 | >2500 | 3.4 |
| 12 | i-Pr | Me | t-butyl | Benzyl | 6 | 274 | -0.9 | |
| 13 | i-Pr | Me | t-butyl | cyclohexyl | 13 | 231 | 2.8 | |
| 14 | i-Pr | Me | t-butyl | Ph | CH2Ph | 9 | 512 | 3.2 |
| 15 | i-Pr | Me | benzyl | Ph | 14 | 544 | 1.3 | |
| 16 | i-Pr | Me | t-butyl | Ph | 14 | >2500 | 1.0 | |
| 17 | i-Pr | Me | benzyl | Ph | 9 | 88 | 1.7 | |
| 18 | i-Pr | Me | benzyl | Ph | 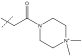 |
17 | >2500 | -0.6 |
| 19 | i-Pr | Me | benzyl | 20 | 540 | 2.0 |
Analysis of crystal structures indicated that the S2 pocket of Der p 1 was shallow in comparison to Cathepsin B (Cat B) and other related proteases. Therefore, as anticipated, we found that a methyl group at P2 gave the best balance of potency and selectivity (compare compounds 5 and 10 in Table 1). In contrast our analysis showed that the S3 pocket of Der p 1 was larger than in Cat B, mainly due to the fact that a threonine residue (Thr74) in Der p 1 is replaced by a bulkier tyrosine residue (Tyr75) in Cat B. Cat S and Cat K also possess bulkier residues (Phe70 and Tyr67, respectively). We therefore reasoned that increasing the steric bulk at P3 would improve the selectivity over these related enzymes. Indeed, we found that switching from benzyl to t-butyl did improve the selectivity whilst maintaining good potency for Der p 1 (compare compounds 6 and 8). Similarly, introducing geminal dimethyl groups on the benzyl group also improved the selectivity over Cat B with only a modest decrease in Der p 1 potency (compare compounds 6 and 9).
By this point in the project we had identified potent Der p 1 inhibitors with good selectivity. Next, we wanted to improve the properties by adding features which either lowered the permeability or increased the binding to lung tissue. Permeability can be reduced by increasing molecular weight and/or PSA, whereas lung tissue retention may be enhanced by combining increasing lipophilicity with a basic or quaternary ammonium group. We found that in P4 a variety of groups were tolerated, including fused rings, basic amines and quaternary amines (e.g. compounds 11, 12 and 13). We also explored the P’ pocket with a series of glycinamide analogues since we anticipated that they could mimic the backbone of the substrate and would have an increased PSA. Fortuitously, we also found that this series had improved selectivity over Cat B (e.g. compound 16).
Dry powder inhalers require stable crystalline compounds and it was pleasing to find that at least 50% of our compounds were crystalline.
In Vivo Efficacy
Having demonstrated that we could obtain potent, selective inhibitors of Der p 1 with a range of physicochemical properties, experiments were carried out to elucidate which features were most strongly correlated with in vivo efficacy.
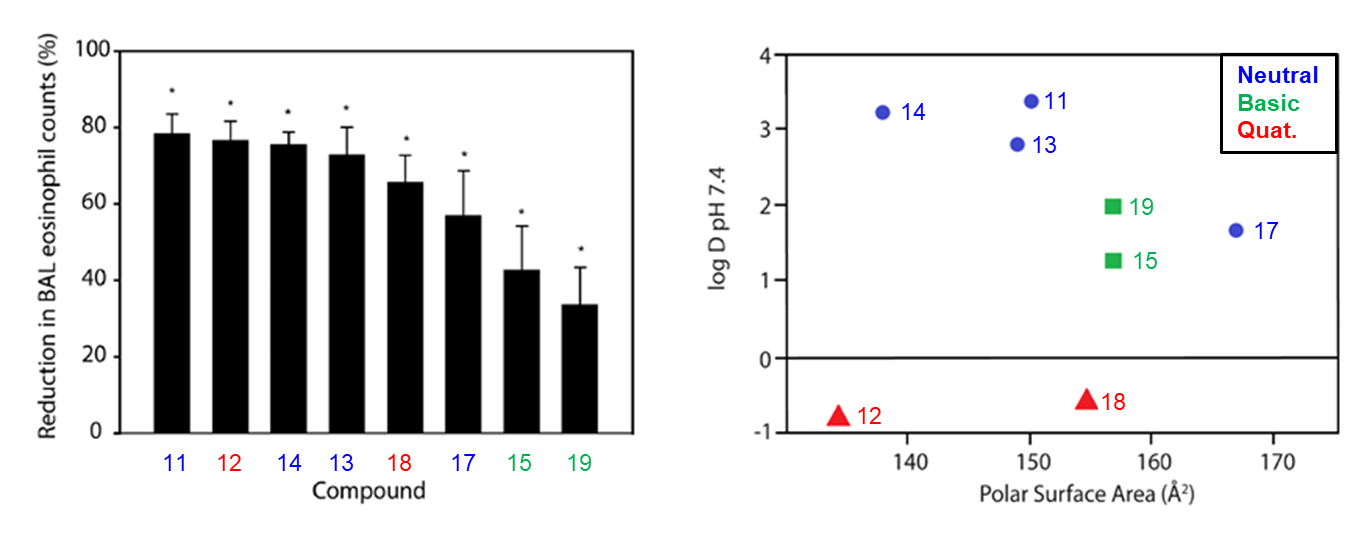
A selection of compounds with similar potencies, but with a range of different log D7.4 and PSA values, were assessed in a disease model (Figure 4). Sensitised Brown Norway Rats were treated with the test compounds by intratracheal aerosolisation, followed by a challenge with a mixture of HDM allergens 2 hours later. The percentage inhibition of eosinophil recruitment was recorded after bronchoalveolar lavage at 48 hours post challenge. More lipophilic compounds showed better efficacy (e.g. compounds 15, 17 and 19). The quaternary ammonium compounds (12 and 18) showed good endurance of action (>6 hours protection from a single dose, data not shown). It was also pleasing to observe that, as anticipated, these compounds had very low levels of systemic exposure (Table 2).
| Compound | F (%) | Cmax 5 mg/kg po dose (nM) | PPB (%) | Free conc Cmax (nM) |
|---|---|---|---|---|
| 12 | 1 | 20 | 61.2 | 8 |
| 18 | 0 | 7 | 63.5 | 3 |
Conclusions
This programme ultimately led to the successful nomination of a candidate drug (the profile of which is shown in Table 3) and funding is now being sought to progress this potentially revolutionary treatment into clinical trials. In addition to the nomination of a CD, the programme identified several developable back-up compounds from chemically and mechanistically distinct series, allowing a robust patent portfolio to be generated.
| Property | Level achieved | |
|---|---|---|
| Potency | On target potency against enzyme | < 10 nM |
| Efficacious dose | 15 μg/kg | |
| Duration of action (rat) | > 6 h | |
| Safety - selectivity | Intrinsic selectivity | > 100-fold |
| Cellular toxicity | > 1000-fold margin | |
| No effect in panel of off-target enzymes and receptors | Clean | |
| Safety - DMPK profile | Systemic exposure Cmax @ 5 mg/kg oral dose | < 100 nM |
| Oral bioavailability (F%) | < 20 % | |
| Plasma protein binding | > 90 % | |
| Hepatic extraction ratio (Eh) | > 0.5 | |
| Pharmaceutical properties | Adequate aqueous solubility | > 50 μM |
| High-melting crystalline form, and no thermal events on heating | > 100 °C | |
| Stable admixed with lactose in solid state | Yes | |
| Stable to micronisation | Yes |
The project was funded by the Wellcome Trust SDD programme and resulted in a publication.4
If you would like to discover more about our services, discuss specific research, or receive a proposal, please contact us or email enquiries@domainex.co.uk
Domainex Expertise
• Integrated Drug Discovery • Medicinal and Synthetic Chemistry • Computational Chemistry • Structure-based Drug Design (SBDD)
• Lead Optimisation • DMPK and ADME • Safety Pharmacology • Protease Drug Discovery • Respiratory Disease Research
References
1. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet, 2017, 390: 1211–59
2. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2016. Geneva, World Health Organisation; 2018
3. The design and synthesis of inhibitors of the cysteinyl protease, Der p I. Billson, J.; Clark, J.; Conway, S. P.; Hart, T.; Johnson, T.; Langston, S. P.; Ramjee, M.; Quibell, M.; Scott, R. K. Bioorg. Med.Chem. Lett., 1998, 8, 993−998
4. The Discovery of Potent, Selective and Reversible Inhibitors of the House Dust Mite Peptidase Allergen Der p 1: An Innovative Approach to the Treatment of Allergic Asthma. Gary K. Newton, Trevor R. Perrior, Kerry Jenkins, Meriel R. Major, Rebekah E. Key, Mark R. Stewart, Stuart Firth-Clark, Steven M. Lloyd, Jihui Zhang, Nicola J. Francis-Newton, Jonathan P. Richardson, Jie Chen, Pei Lai, David R. Garrod and Clive Robinson., J. Med. Chem., 2014, 57(22):9447-62
Start your next project with Domainex
Contact one of our experts today