- About
-
Solutions
-
Services
-
Biosciences
- PureDisc™ Enabled Membrane Protein Production
- Assay Development and Transfer
- Biochemistry / Enzymology
- Biomarker Services
- Biophysics
- Cellular Biology
- Compound screening and cascade design
- Cryo-EM
- Fibrosis Assays
- Project Management and Consultancy Services
- Protein Expression
- Structural Biology
- X-ray Crystallography
- Chemistry
- Integrated Drug Discovery
- Computer Aided Drug Design
- Hit ID and Screening Services
- DMPK Services
-
Biosciences
- Target Classes and Modalities
- Therapeutic Areas
-
A-Z
- A
- B
- C
- D
- E
- F
- G
- H
- I
- K
- L
- M
- N
- O
- P
- R
- S
- T
- V
- X
-
Services
- Library
- News & Events
- Careers
- PureDisc™
MAP4K4: Kinase inhibitors for cardioprotection following heart attacks
Domainex worked with Imperial College to design novel, potent, highly selective, small-molecule inhibitors of MAP4K4, with potential cardio-protective drug properties.

Challenge
Heart disease is the paramount global cause of death and disability. Although the death of heart muscle cells (cardiomyocytes) plays a causal role, and hence a cardio-protective drug would be a logical treatment, no existing therapeutics target the intracellular pathways responsible for cardiomyocyte death. Progress to this end has been hampered by a lack of preclinical human validation of prospective drug targets.1,2
Solution
Mitogen-Activated Protein Kinase Kinase Kinase Kinase 4 (MAP4K4), a serine-threonine kinase which activates the JNK signalling pathway, is activated in failing human hearts and relevant rodent models. Working in collaboration with Professor Michael Schneider and his team at Imperial College, we used human induced-pluripotent-stem-cell-derived cardiomyocytes (hiPSC-CMs) (Figure 1a) and MAP4K4 gene silencing (Figure 2), to demonstrate that cardiomyocyte death induced by oxidative stress requires MAP4K4.
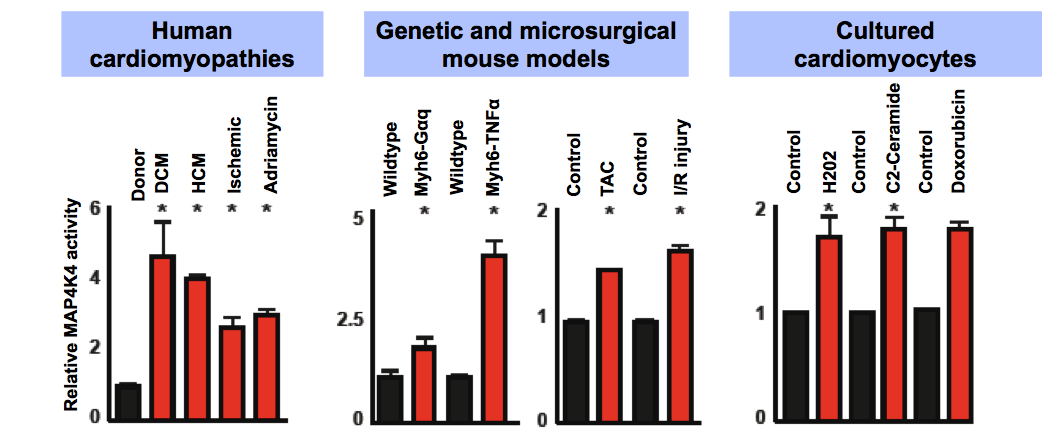
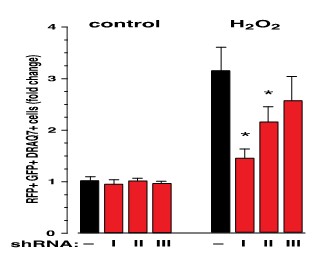
Figure 2: Potent short hairpin RNAs (shRNAs) conferred protection against H2O2
Therefore, we postulated that an inhibitor of MAP4K4 would be able to suppress human cardiac cell death. To maximise the clinical benefit of a MAP4K4 inhibitor, rapid onset would be required following myocardial infarction, and we believed this would be best achieved by delivering an intravenous bolus of drug followed by a maintenance infusion. In order to achieve this, a drug with high aqueous solubility would be essential.
Hit Identification
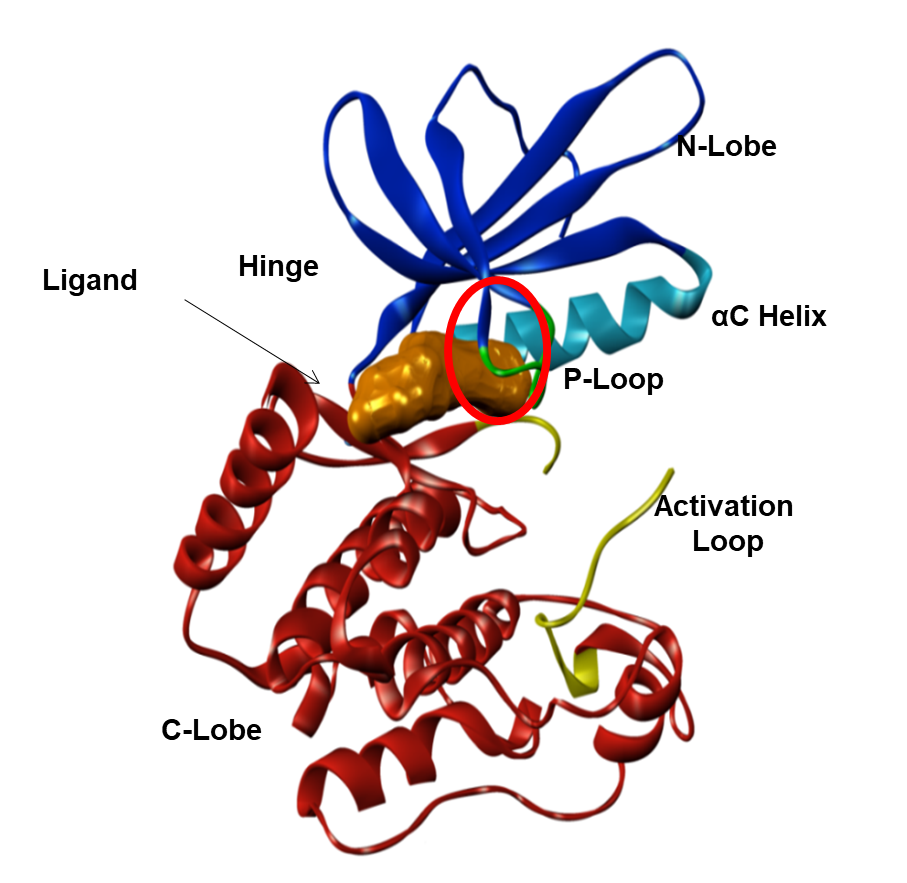
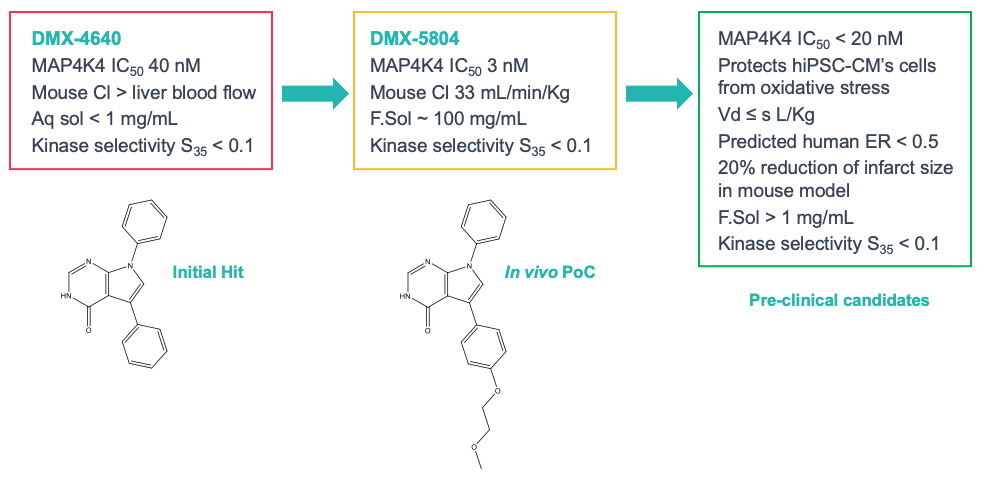
An empirical screen was carried out against human MAP4K4 using about 1800 bioactive compounds. The best hit compounds were then analysed and a consensus pharmacophore was identified using a molecular field-based approach.3 This pharmacophore was used to carry out a ligand-based virtual screen to identify related compounds for biochemical testing. The starting point for medicinal chemistry arising from these CADD studies, DMX-4640 (Figure 3), had good potency (40nM) for MAP4K4 and good selectivity over other protein kinases in a broad selectivity panel, but its PK properties and solubility needed optimisation. We were able to rationalise the selectivity of DMX-4640 by docking it into a crystal structure of MAP4K4 (Figure 3), which revealed an unusual protein conformation with a “folded” P-Loop motif. This feature was exploited in order to further refine the selectivity of this chemical series.
Compound Optimisation
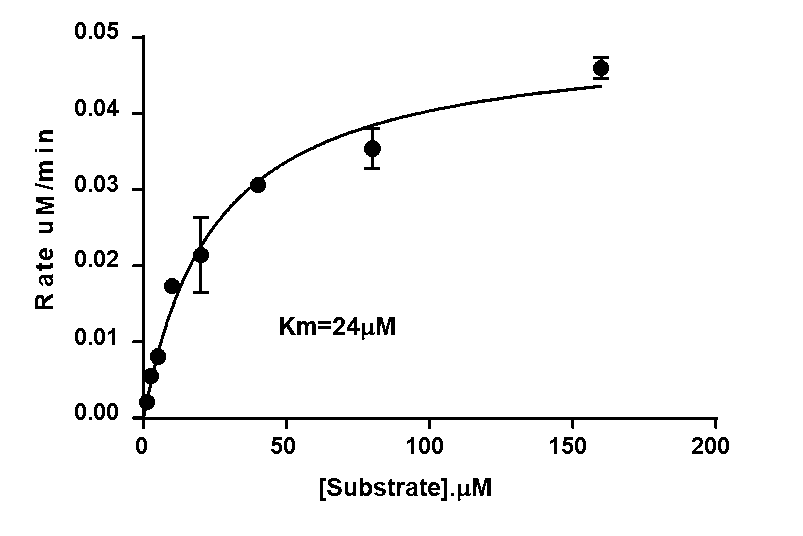
Domainex developed biochemical assays for MAP4K4 and six key off-target kinases. These were run as a selectivity panel, where each kinase was tested using [ATP] at its Km value (Figure 4). Within just 6 weeks of the client authorising the work, the Domainex team had completed the assay development and started routine screening.
| Kinase | ATP Km (μM) | [MBP].μM |
|---|---|---|
| MAP4K4 | 24 | 1 |
| Family Mem 1 | 26 | 1 |
| Family Mem 2 | 104 | 1 |
| Off Target 1 | 18 | 1 |
| Off Target 2 | 94 | 1 |
| Off Target 3 | 11 | 20 |
| Off Target 4 | 16 | 1 |
Starting with DMX-4640, we embarked upon a programme of rational drug design with the aims of improving potency, metabolic stability and aqueous solubility, whilst maintaining the excellent selectivity of the template. For example, introducing a 2-methoxyethoxy group onto one of the benzene rings gave DMX-5804, a compound with a significantly greater water solubility and a reduced metabolic clearance. Further structural modifications led to the design of other compounds with excellent properties, that were fully consistent with those required for clinical candidates (Figure 5).
Target Validation
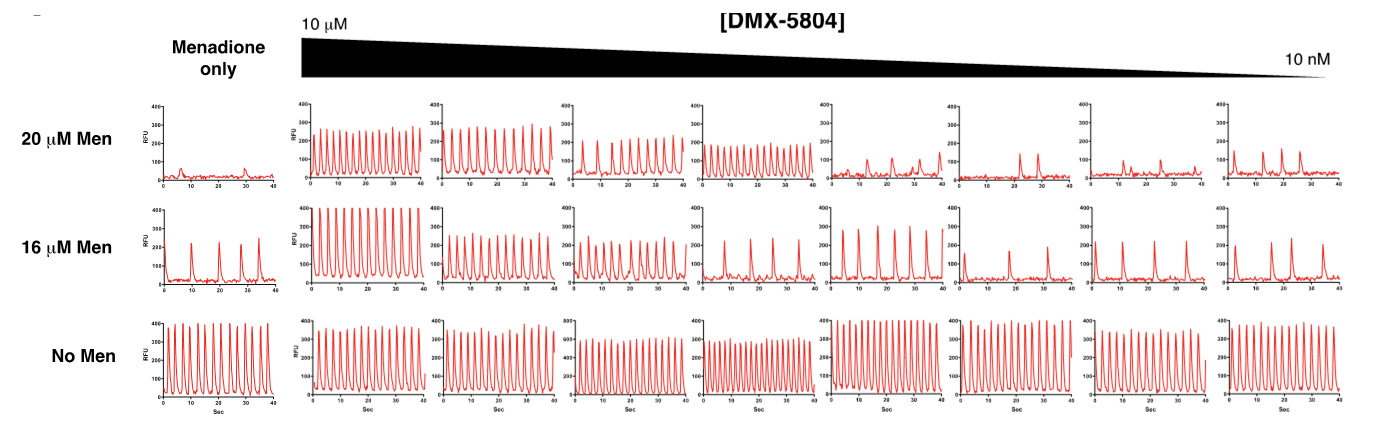
Given its potency and selectivity, DMX-5804 was investigated for its ability to preserve key aspects of human cardiomyocyte function after oxidative stress. Calcium cycling, a hallmark of the cardiac phenotype, is susceptible to redox- and phosphorylation-dependent abnormalities.4 So, to determine whether MAP4K4 inhibition might preserve calcium homeostasis, vCor.4U hiPSC-CMs were challenged with menadione (vitamin K3, which induces intracellular reactive oxygen species through quinone redox cycling5) in the presence of a fluorescent intracellular calcium indicator (Figure 6). This showed that calcium cycling was sensitive to oxidative stress even at sub-lethal concentrations of menadione. Furthermore, DMX-5804 dose-dependently protected cardiomyocyte function from menadione stress, while showing no toxic effect on cardiomyocyte function. Protection was maintained for up to 96 hours, despite the presence of menadione throughout.
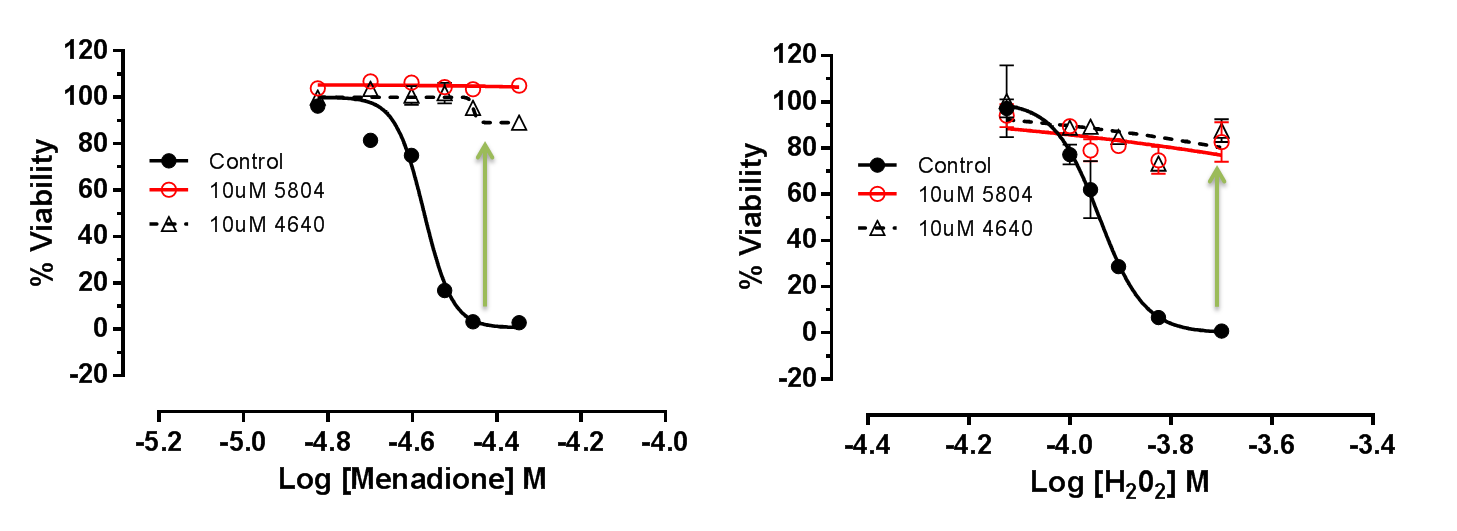
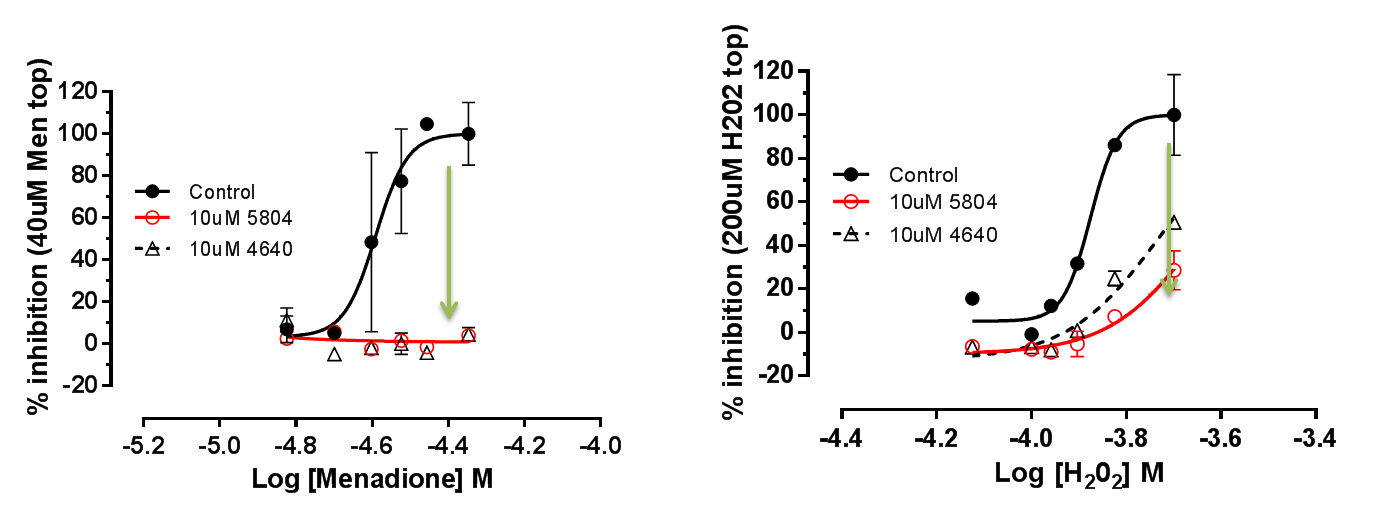
We demonstrated that MAP4K4 inhibition confers protection in hiPSC-CMs (Figure 7). Cardiac Troponin release is a clinical biomarker for myocardial infarction, and we showed that DMX-5804 could reduce this release (Figure 8).
To test whether MAP4K4 target validation and compound development in hiPSC-CMs might predict success in a whole-animal context, mice undergoing experimental myocardial infarction were treated with DMX-5804 or the vehicle control (Figure 9). Treatment was begun either 20 min prior to ischemia or 1 hour after reperfusion injury, the latter having greater clinical relevance. Exposure was demonstrated to be in excess of the anticipated EC50 from cellular protection assays (Figure 10). In both cases, ischemia-reperfusion injury in mice was reduced by >50%.
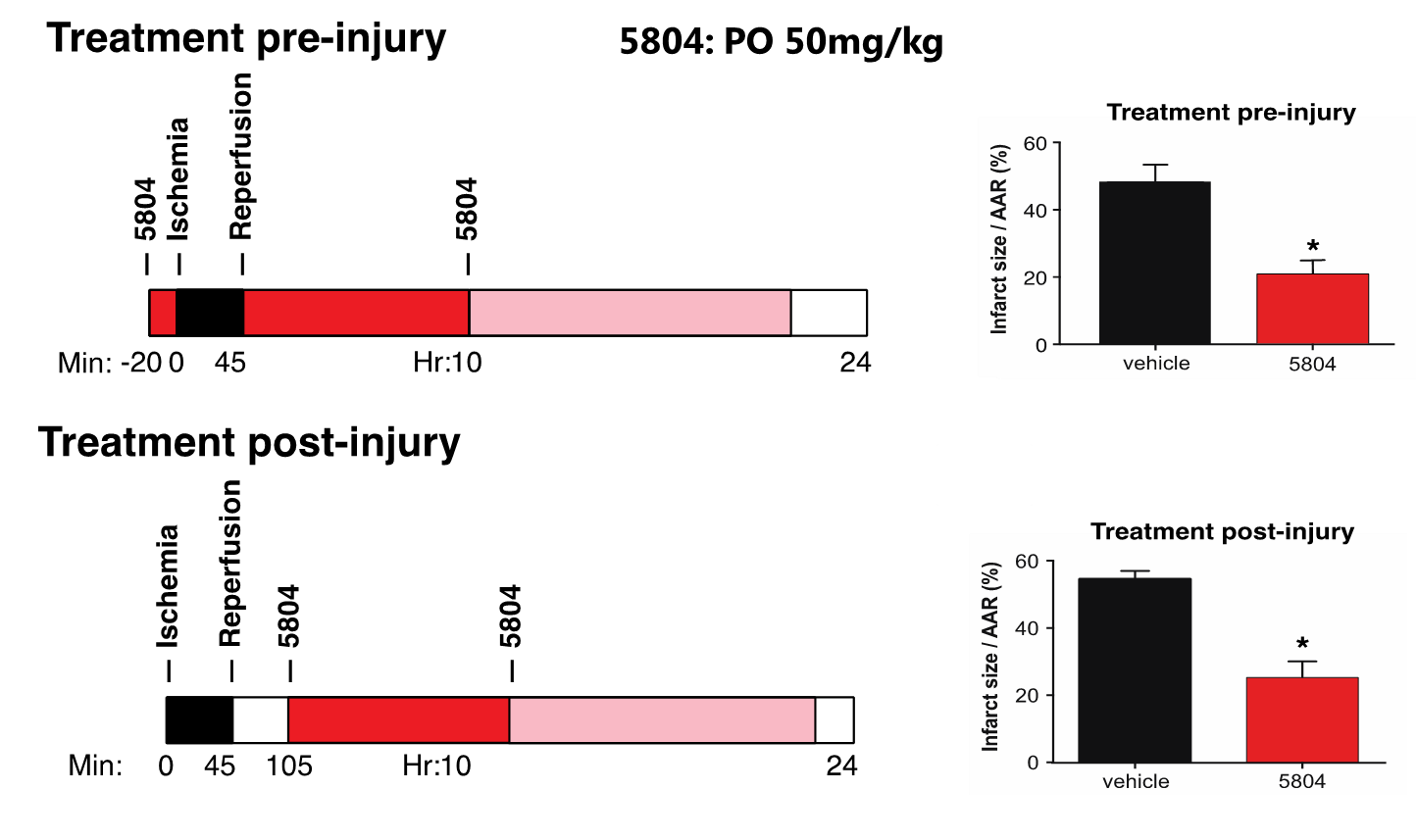
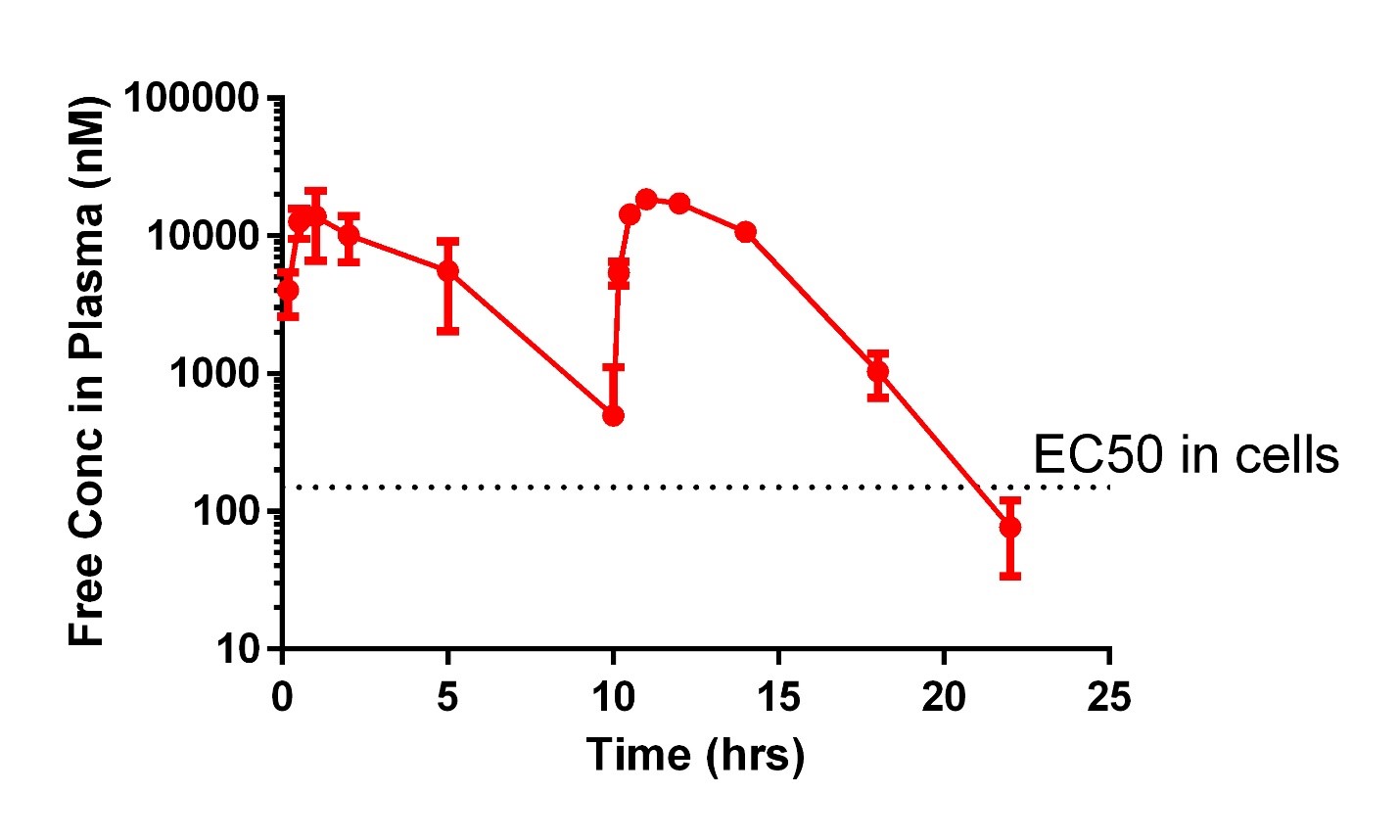
Figure 10: Exposure of DMX-5804 in mice was in excess of the anticipated EC50 from cellular protection assays after dosing at 50mg/kg
Conclusions
Using hiPSC-CMs as the most relevant platform for gene silencing and drug discovery, we have designed small-molecule inhibitors of MAP4K4. DMX-5804, a novel, potent, highly selective, small-molecule inhibitor of MAP4K4, was identified for proof of concept studies. DMX-5804 rescues cell survival and function in hiPSC-CMs and reduces ischemia-reperfusion injury in mice by >50%. The solubility and pharmacokinetic properties of DMX-5804 were insufficient for a human drug candidate in acute ischemic injury, where rapid intravenous infusion is desired. However, we have been able to further optimise this series and have identified a number of compounds with suitable properties.
This research was funded by the British Heart Foundation, the Medical Research Council and the Wellcome Trust.
This work resulted in a publication.6
Domainex Expertise
• Integrated Drug Discovery • Computational Chemistry • Medicinal and Synthetic Chemistry • Assay Development • Biochemistry • Cell Biology
• Lead Optimisation • DMPK and ADME • Safety Pharmacology • X-ray Crystallography • Kinase Drug Discovery • Cardiovascular Research
References
1. Cardiovascular drug development: is it dead or just hibernating? Fordyce, C.B., Roe, M.T., Ahmad, T., Libby, P., Borer, J.S., Hiatt, W.R., Bristow, M.R., Packer, M., Wasserman, S.M., Braunstein, N., et al. J. Am. Coll. Cardiol., 2015, 65, 1567–1582.
2. Cardiovascular drug discovery: a perspective from a research-based pharmaceutical company. Gromo, G., Mann, J., and Fitzgerald, J.D. Cold Spring Harb. Perspect. Med. 2014, 4, a014092.
3. High content pharmacophores from molecular fields: a biologically relevant method for comparing and understanding ligands. Cheeseright, T.J., Mackey, M.D., and Scoffin, R.A. Curr. Comput. Aided Drug Des., 2011, 7, 190–205.
4. The structural basis of ryanodine receptor ion channel function. Meissner, G. J. Gen. Physiol. 2017, 149, 1065–1089.
5. Anticancer vitamin K3 analogs: a review. Badave, K.D., Khan, A.A., and Rane, S.Y. Anticancer. Agents Med. Chem. 2016, 16, 1017–1030.
6. MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Lorna R. Fiedler, Kathryn Chapman, Min Xie, Evie Maifoshie, Micaela Jenkins, Pelin Arabacilar Golforoush, Mohamed Bellahcene, Michela Noseda, Do¨rte Faust, Ashley Jarvis, Gary Newton, Marta Abreu Paiva, Mutsuo Harada, Daniel J. Stuckey, Weihua Song, Josef Habib, Priyanka Narasimham, Rehan Aqil, Devika Sanmugalingam, Robert Yan, Lorenzo Pavanello, Motoaki Sano, Sam C. Wang, Robert D. Sampson, Sunthar Kanayaganam, George E. Taffet, Lloyd H. Michael, Mark L. Entman, Tse-Hua Tan, Sian E. Harding, Caroline M.R. Low, Catherine Tralau-Stewart, Trevor Perrior and Michael D. Schneider; Cell Stem Cell, 2019, 24 (4), 579-591.
Press Releases
Start your next project with Domainex
Contact one of our experts today