- About
-
Solutions
-
Services
-
Biosciences
- PureDisc™ Enabled Membrane Protein Production
- Assay Development and Transfer
- Biochemistry / Enzymology
- Biomarker Services
- Biophysics
- Cell-Based Assays
- Compound screening and cascade design
- Cryo-EM
- Fibrosis Assays
- Project Management and Consultancy Services
- Protein Expression
- Structural Biology
- X-ray Crystallography
- Chemistry
- Integrated Drug Discovery
- Computer Aided Drug Design
- Hit ID and Screening Services
- DMPK Services
-
Biosciences
- Target Classes and Modalities
- Therapeutic Areas
-
A-Z
- A
- B
- C
- D
- E
- F
- G
- H
- I
- K
- L
- M
- N
- O
- P
- R
- S
- T
- V
- X
-
Services
- Library
- News & Events
- Careers
- PureDisc™
TBK1/IKKε: Kinase inhibitors for the treatment of interferonopathies
TANK-binding kinase 1 (TBK1) and IkappaB kinase epsilon (IKKε) have been validated as novel drug targets in inflammatory diseases including interferonopathies, such as systemic lupus erythematosus.
Challenge
Systemic lupus erythematosus (SLE) is an incurable disease of the immune system that principally affects the kidneys and skin but can also damage other major organs such as the heart, lungs and brain. Typical symptoms are joint and muscle pain and extreme tiredness, often accompanied by rashes, ulceration, and headaches. Anti-inflammatory drugs, notably steroids, can be used to manage the disease and this has improved the quality of life of SLE patients, although their life expectancy is still lower than that of the healthy population. Heart and kidney failure are the most common causes of death in SLE patients. In 2012 it was estimated that at least five million people worldwide, have a form of lupus.1
Aberrant activation of interferon signaling, and the upregulation of Interferon Stimulated Genes (ISG), are amongst the hallmarks of SLE. Two kinases, TBK1 and IKKε, are known to orchestrate the induction of interferons and activation of inflammatory genes; therefore, an inhibitor of these enzymes should lead to a disease-modifying effect in SLE and other related interferonopathies such as Sjögren’s syndrome, and systemic sclerosis.
No inhibitors of TBK1 and IKKε with a good ADMET profile are to be found in the literature, and this was expected to be the main challenge for the programme.
Solution
Hit Identification phase
In order to identify hit compounds, Domainex initially conducted a fragment screen using a focused library of low molecular weight compounds selected to contain motifs that might bind to the so-called ‘hinge’ that is found between the N and C terminal lobes of the conserved catalytic domain of protein kinases. The library was screened at high concentration using a HTRF biochemical assay. A homology model of IKKɛ was generated by Domainex’s computational chemistry team and the fragment hit shown in Figure 1 below was docked into this model (Figure 2) and then progressed into a structure-guided hit elaboration phase.

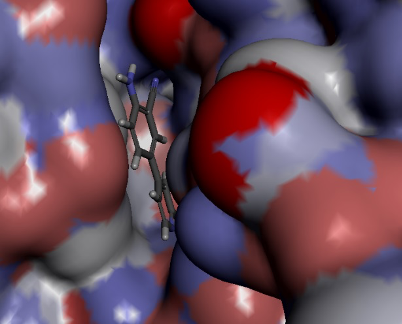
Hit Elaboration Phase
Replacing the 4-amino group with a pyrrolidine led to a significant increase in potency, while maintaining the ligand efficiency (LE) above 0.4. Introducing aryl or heteroaryl amino substitution onto the pyridyl ring also led to an increase in potency (~10 fold), and changing the pyridyl group to a pyrimidine enhanced both the potency and LE by removing the steric clash between the periplanar hydrogen atoms of the two pyridine rings (Figure 3).
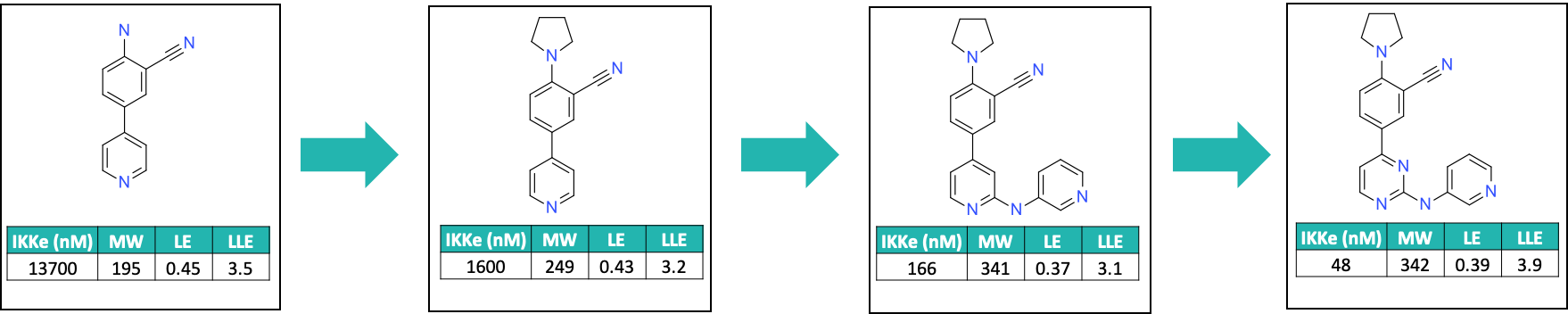
It is interesting to note that despite our best efforts to optimise the original hit in a number of different directions, the preferred chemotype that emerged from these studies was an aminopyrimidine, which is a common motif found in kinase inhibitors including, for example, imatinib. However, the nitrile group was able to provide selectivity and novelty that differentiated these compounds from other inhibitor series.
At this stage, the potency and selectivity of the compounds was good, but more work needed to be done to improve the ADME profile, particularly the aqueous solubility, metabolic stability and permeability. The strategy adopted to address these issues was to block key sites of metabolism and lower the logD.
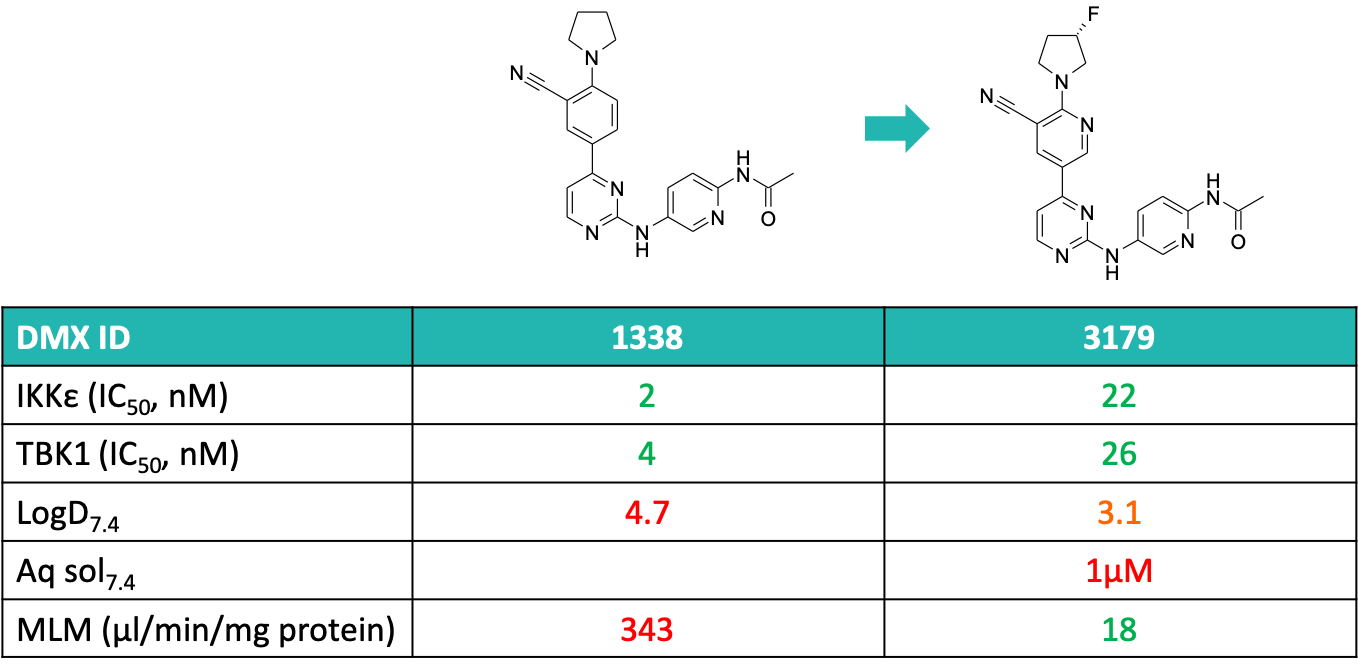
Optimisation
The first breakthrough was the addition of a fluorine to the 3-position of the pyrrolidine ring (see Figure 4) to improve metabolic stability. Then in order to lower the logD, the phenyl core was replaced with a number of heterocyclic rings, including all three isomeric pyridines. The pyridine isomer shown had the best properties, and this was used as a template for further modification of the 2-substituent on the distal pyridine ring in order to further lower the logD and improve the aqueous solubility. This led to DMXD-011, which exhibited excellent drug like properties (see Figure 5).

Animal disease model
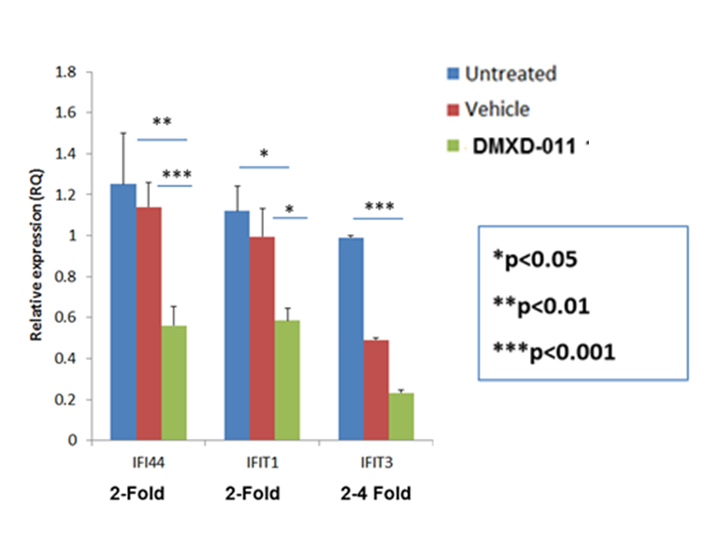
Hasan et al2 reported that TBK1 plays a role in familial chilblain lupus which contains D18N mutation. In a collaboration with Professor Fred Perino at Wake Forest Medical School, we showed that DMXD-011 reduced expression of Interferon Stimulated Genes (ISG) in D18N mutant mice (see Figure 6).
Ex vivo human studies
In another collaboration with Marjan Versnel (Erasmus Uni), we carried out an ex vivo human study to examine the effect of DMXD-011 on levels of cytokines such as IFNα and IP-10 following imiquimod (IMQ) stimulation of TLR7, and on the spontaneous expression of ISG in PBMCs. Blood samples were taken from healthy volunteers, and from patients with SLE, Sjögren’s syndrome, and systemic sclerosis (SSc).
DMXD-011 dramatically reduced the effect of the IMQ stimulation on cytokine levels (Figure 7).
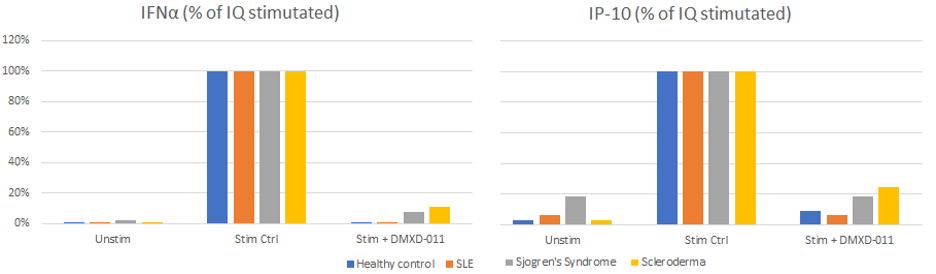
Versnel also measured the relative mRNA levels of exemplar ISG in PBMCs sourced from interferonopathy patients before and after 5h treatment with DMXD-011 (Figures 8 & 9).
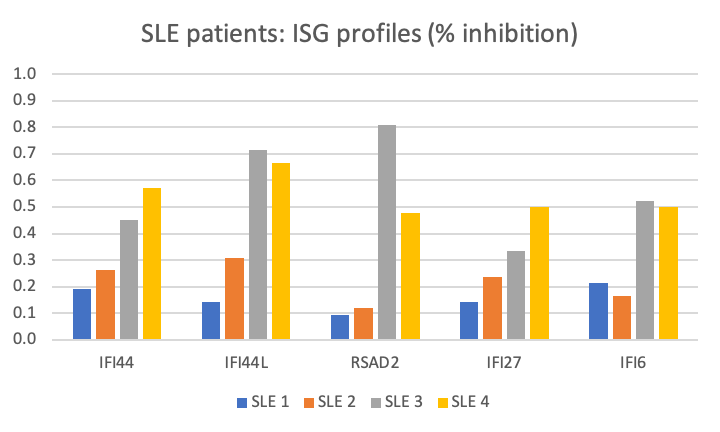
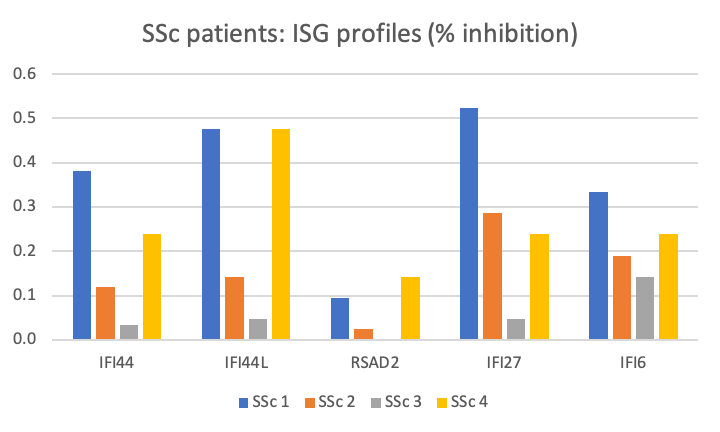
Conclusions
Domainex has identified a drug candidate DMXD-011, a first-in-class small-molecule orally-bioavailable inhibitor of TBK1/IKKε, and based on the animal and human ex vivo data, it is currently progressing this compound as a disease-modifying therapy for systemic lupus erythematosus, and related interferonopathies. DMXD-011 has also demonstrated efficacy in a range of other rodent inflammatory models including LPS challenge and collagen-induced arthritis.
If you would like to discover more about our services, discuss specific research, or receive a proposal, please contact us or email enquiries@domainex.co.uk.
Domainex Expertise
• Integrated Drug Discovery • Hit Identification • Fragment Screening • Medicinal and Synthetic Chemistry • Hit-to-lead • Lead Optimisation
• Structure-based Drug Design (SBDD) • Computational Chemistry • Assay Development • Biochemistry • DMPK and ADME • Safety Pharmacology
References
1. GfK Roper. (2012). Lupus Awareness Survey for the Lupus Foundation of America [Executive Summary Report]. Washington, DC
2. Trex1 regulates lysosomal biogenesis and interferon independent activation of antiviral genes. Maroof Hasan, James Koch, Dinesh Rakheja, Asit K. Pattnaik4, James Brugarolas, Igor Dozmorov, Beth Levine, Edward K. Wakeland, Min Ae Leekirsch, and Nan Yan; Nat Immunol., 2013,14(1), 61–71.
Start your next project with Domainex
Contact one of our experts today