By the Domainex Synthesis Group (Alicia Galván Álvarez, Hugh Tawell, Tenin Traore, Andrew Jones, David Gibson, Andrea Bombana, James Sharpe, Venu Komanduri, Fergus Preston, Brahmam Medapi and Robyn Presland)
In this edition of our blog series, we have focused on the following two recent publications, which describe highly useful synthetic transformations:
- C−F bond activation enables synthesis of aryl difluoromethyl bicyclopentanes as benzophenone-type bioisosteres. Xiaheng Zhang et. al., Nat. Commun., 2024, 419, 15,
- Regiodivergent Arylation of Pyridines via Zincke Intermediates. Michael F. Greaney et. al., Angew. Chem. Int. Ed., 2024.
Benzophenone Bioisosteres
Bioisosteric replacement plays a crucial role in the development of new drug candidates by modulation of bioavailability and metabolic stability. Small-ring cage hydrocarbons (such as bicyclo- [1.1.1]pentane (BCP)) remain a popular bioisostere for benzene rings due to improved pharmacokinetic properties. Whilst benzene bioisosteric replacements have been extensively explored (including various substitution patterns), replacements for benzophenone functionality (an important scaffold) remain relatively under-explored. To this end, Zhang and co-workers report the synthesis and use of difluoromethyl BCP arenes as a replacement for a benzoyl group.1
![Small-ring cage hydrocarbons (such as bicyclo- [1.1.1]pentane (BCP)) remain a popular bioisostere for benzene rings. Whilst, Zhang and co-workers report the use of difluoromethyl BCP arenes as a replacement for a benzoyl group.](/sites/default/files/inline-images/Screenshot%202024-02-19%20at%2011.30.32.png)
Previous methods to access these structures rely on the use of difluoroalkyl bromides. This approach lacks generalisability, in part due to the inaccessibility/cost of privileged difluoroalkyl bromides. To address this issue, the authors utilise a photochemical C-F bond activation strategy from readily available trifluoromethyl arenes. The corresponding difluorobenzylic radical can then be trapped by [1.1.1]propellane to afford a BCP radical which can then partake in hydrogen-atom transfer (HAT) or borylation to give the desired aryl difluoromethyl bicyclopentane (ADB) scaffold.
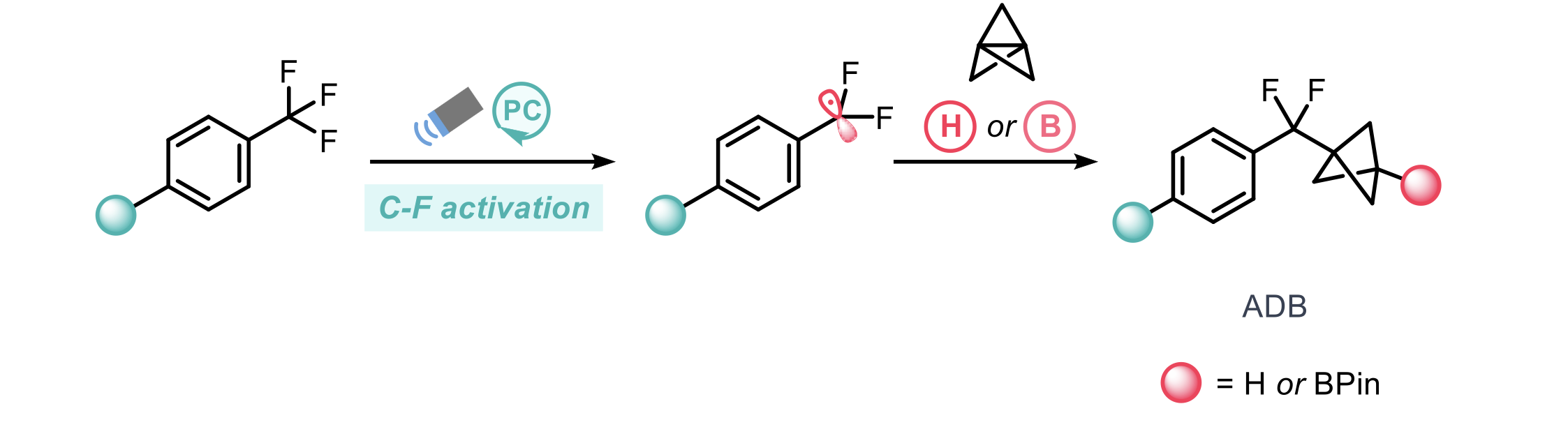
Initial efforts demonstrated product formation (albeit in low yields) when employing common photocatalysts under blue light (427 nm LEDs). This led to the design of a bespoke o-phosphinoarylamine (PN) to drastically improve reactivity, for some cases, 2-(diphenylphosphino)biphenyl amine (PBN) was used instead. The HAT reagent used was γ-terpinene.
The scope of the defluorinative coupling when trapping with the HAT reagent demonstrated good generality over a range of substrate classes providing the desired ADBs in moderate to good yields across >45 examples, including heteroaromatics, bicyclic arenes and drug-like analogues. Further to this, the methodology could also be extended to the construction of bicyclo[3.1.1]heptanes and [4.1.1]bicyclooctane scaffolds.
![Top: Defluorinative coupling with [1.1.1]propellane; Bottom: Deflorinative 3-component coupling.](/sites/default/files/inline-images/Screenshot%202024-02-19%20at%2011.32.21.png)
To further demonstrate the potential of ADB scaffolds in a medicinal chemistry setting, bioisosteres of the established adiponectin receptor agonist ‘AdipoRon’ were synthesised via a 2-step sequence (condensation followed by photocatalytic defluorinative coupling). Interestingly, the resulting difluoro [1.1.1]-BCP analogue was shown to be metabolically stable, albeit with decreased membrane permeability (Caco-2). These promising initial results demonstrate the capability of the ADB scaffold as a bioisostere for a benzophenone core.
Pyridine Regiodivergent Arylation
(Hetero)Aryl-substituted pyridines are a valuable motif, present in a number of pharmaceuticals and natural products. Despite this, their preparation via the arylation of pyridine remains challenging because it often lacks selectivity and requires an excess of pyridine to be used. An alternative to the direct functionalisation of pyridines is to activate the pyridine nitrogen and treat the resulting pyridinium intermediate with a nucleophile to yield a dearomatised Zincke imine. The Zincke imine is more amenable to functionalisation and can be re-aromatised to the functionalised pyridine.
In their recent publication, Greaney et al.2 report that Zincke imines, which are formed by the treatment of pyridine with triflic anhydride and a secondary amine undergo regiodivergent arylation. It was found that the palladium catalysed arylation of Zincke imines with aryl iodides was selective for the 4-position, whereas metal free arylation with diaryliodonium salts was selective for the 5-position. Recyclisation of the arylated Zincke imines yields the corresponding 4- and 5- substituted pyridines respectively.
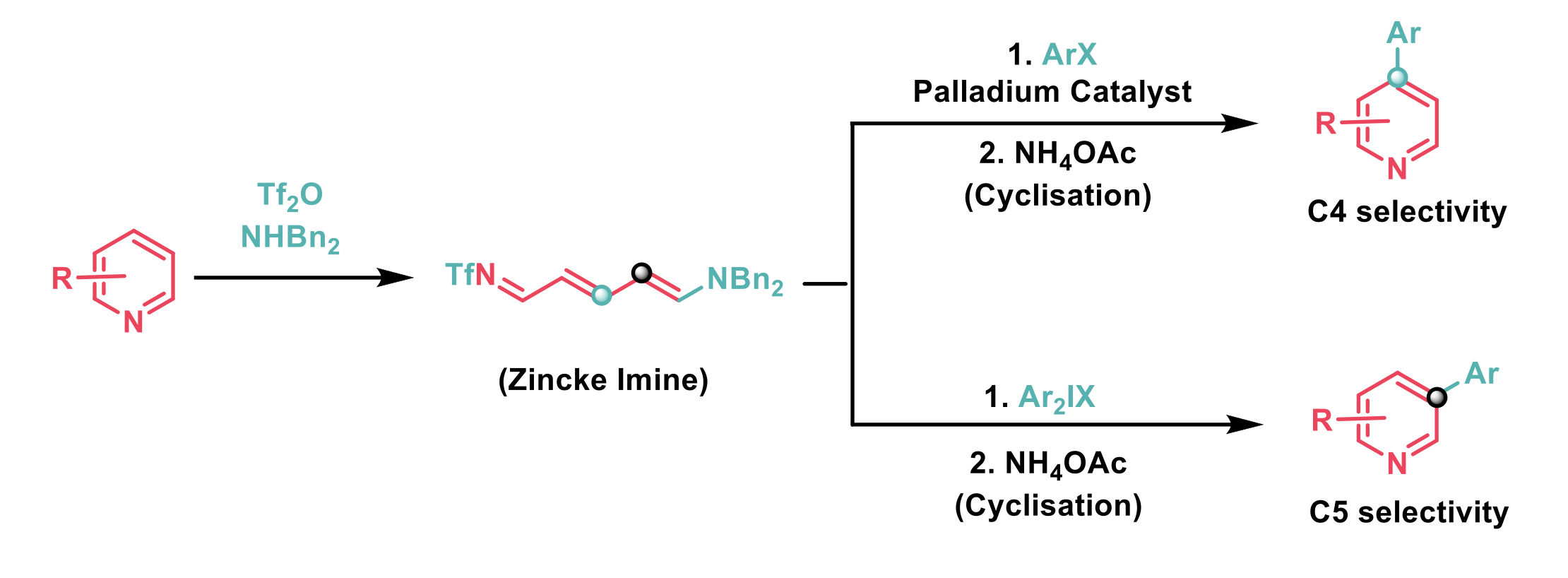
A range of aryl iodides containing alkyl, aryl, halo, alkoxy, trifluoro, and ester functional groups successfully arylated the Zincke imine in the palladium catalysed reaction. Aryl bromides were also compatible but gave poorer yields. Both para- and meta-substitution on the aryl iodide coupling partner were well tolerated, although ortho-substitution was not well-tolerated. The reaction was found to be very selective for the 4-position and even when this position was blocked very little of the other possible regioisomers were observed. Pleasingly, all three steps could be conveniently conducted in one-pot with a single solvent switch. The authors postulate that under the reaction conditions the Zincke imine could undergo hydrolysis to form the corresponding ketone, and that the formed ketone could undergo a Heck-type reaction with the aryl iodide. Although the necessity of a carbonate base in the reaction may indicate a C-H activation pathway. Mechanistic studies are underway.
A short substrate scope study of the electrophilic arylation with diaryliodonium salts demonstrated that halo-substitution in the diaryliodonium salt was tolerated and that substrate substitution adjacent to the arylation site in pyridine was also tolerated.
We hope you found this blog interesting. Our next review will be available soon, but in the meantime, get in touch to find out how we can help solve your synthetic chemistry challenges.