By Anna Hopkins (Group Leader, Chemistry)
Fragment based drug discovery, coupled with structure-based design is a key weapon in the arsenal of modern drug discovery. Astex have been a pioneer in this field, and their recently published paper on the discovery of allosteric inhibitors of SH2 containing protein tyrosine phosphatase-2 (SHP2) demonstrates clever synergy between computational profiling, X-ray crystallography and medicinal chemist design1.
SHP2 has been indicated in various cancers, including KRAS mutant cancers and RTK-driven cancer cell lines. Historically, protein phosphatases have been challenging due to the nature of their highly conserved active site, resulting in ligands with poor selectivity and PK properties. Recently, allosteric sites have been identified which can stabilise an auto-inhibited ‘inactive’ form of the enzyme, and Astex planned a campaign to identify fragments that could exploit this pocket.
Implementation of Astex’s PyramidTM platform using both ligand-observed NMR and X-ray crystallography lead to 88 validated fragments (5.3% hit rate). Fragments were observed binding to multiple pockets on the protein, however most hits (83 fragments) bound to a tunnel-like site at the interface between the C-SH2 domain, the PTP domain and the linker region between the C-SH2 and N-SH2 domains. Although a novel binding site between the interface of the N-SH2 and PTP domains (Domain Interface 1) was briefly investigated, most of the work described focused on fragments at the tunnel site.
The first step for the team investigating the tunnel site was solvent mapping molecular dynamics to probe the energetics of the water network within the binding pocket. This analysis was able to categorise waters into stable, less stable, and unstable and thus provided a road map for areas of growth likely to yield improved potency. Three areas of interest were identified, and further analysis of the electrostatic surface of the pocket allowed a strategy for optimisation to be developed.
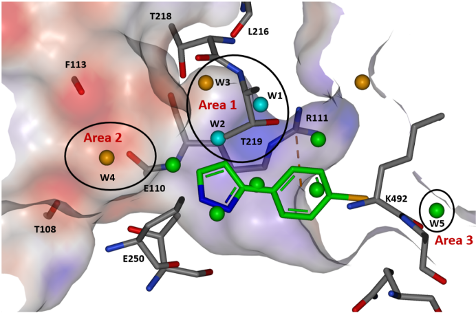
Figure 1: View of the tunnel site in full-length SHP2 and modelled water sites overlaid with binding pose of fragment hit 3. Reproduced with permission from Astex Pharmaceuticals1
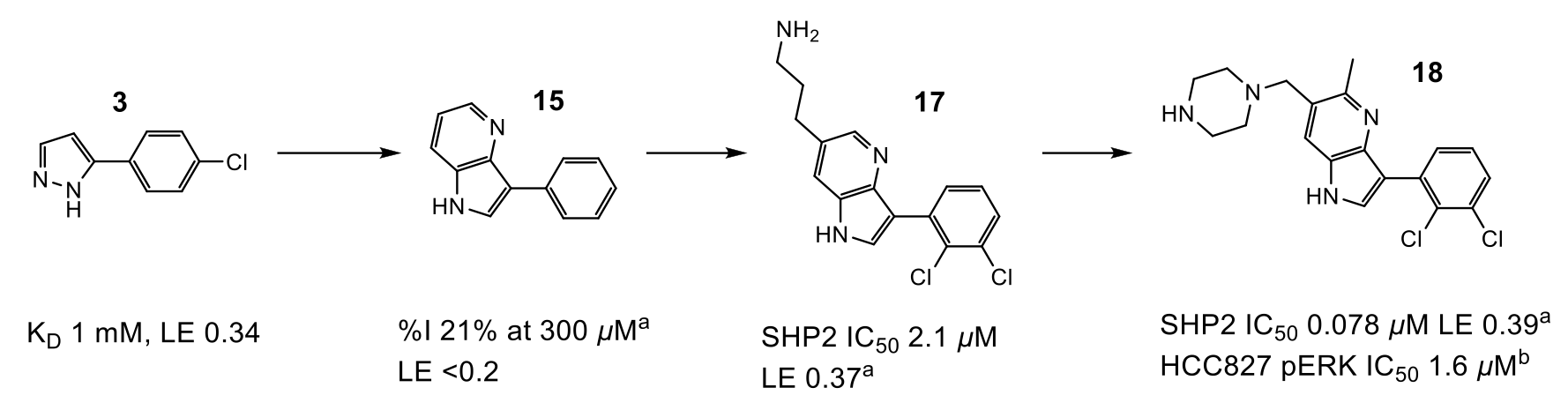
Figure 2: Progression of compound design. aIC50 or %I values determined by fluorescence assay. bMeasurement of compound effect on pERK levels in HCC827 cells
Optimisation of fragment 3 began with growth to bicycle 15, which better filled the pocket and utilised a pyridyl nitrogen atom to replace a stable water (W2) observed in the site. The C-6 position of the pyrrolopyridine ring proved the best vector to explore electronegative Area 2 (Figure 1), and subsequent protonated amine side chains (both acyclic (17) and constrained) demonstrated the expected improvement in potency. Additionally, a polarised methyl group at the C-5 position yielded a 5-fold increase in activity (18). This compound had activity in a HCC827 pERK cell assay (1.6 µM) and an antiproliferative effect in the same cell line.
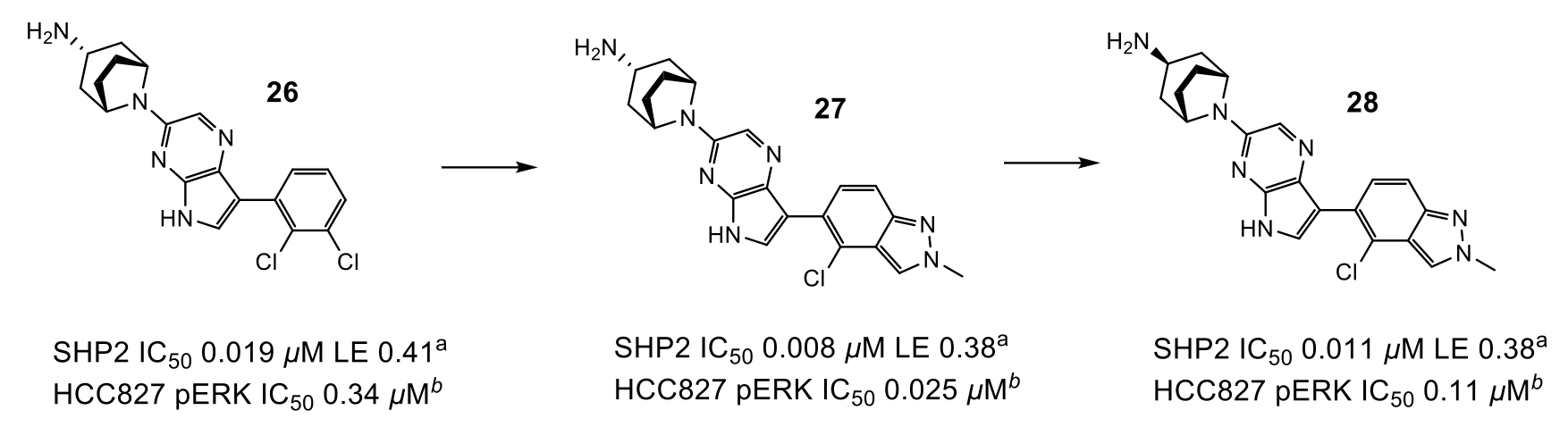
Figure 3: Progression to lead compound. aIC50 or %I values determined by fluorescence assay. bMeasurement of compound effect on pERK levels in HCC827 cells
To optimise the interactions of the basic amine in Area 2, as well as move away from a di-basic structure, the researchers mined the Cambridge Structural Database (CSD) to find several examples of mono-basic tropanes which adopted the desired conformation as predicted from X-ray structure. QM calculations suggested that these would preferentially adopt the optimum geometry and implementation of this group gave the desired uplift in potency, particularly when coupled with a more electron-deficient pyrrolopyrazine ring (26).
The final area of investigation grew from the phenyl ring towards an unstable water in Area 3 (Figure 1). Unfortunately, displacement of this water did not produce the expected uplift in potency, and a smaller group was selected, which exhibited an uplift in potency but did not displace the water.
Combination of efforts in Areas 2 and 3 led to compound 27 which exhibited 25 nM activity in the pERK cell assay. Unfortunately, compound 27 had low oral bioavailability due to poor permeability, likely driven by transporter mediated efflux (Caco-2 0.1 x 10-6 cm/s, ER 14). Surprisingly, endo-tropane 28 showed similar activity (despite not fully engaging residues in Area 2) and low efflux (Caco-2 5.4 x 10-6 cm/s, ER <1), leading to a highly bioavailable compound that was suitable for PK/PD evaluation. Dosing 28 to mice bearing HCC827 tumour xenografts demonstrated a decrease in pERK and pRSK and inhibition of tumour growth in a dose-dependent manner.
In conclusion, Astex have described an elegant example of fragment-to-lead discovery against the SHP2 enzyme. Interrogation of the X-ray structure by electrostatics and solvent mapping analysis, together with conformational analysis were key tools in guiding the optimisation strategy.
If you would like support with a fragment based drug discovery approach, please get in touch to speak to one of our experts.
Reference:
- Day et al. Fragment-Based Discovery of Allosteric Inhibitors of SH2 Domain-Containing Protein Tyrosine Phosphatase-2 (SHP2). J Med. Chem., 2024, 67, 6, 4655-4675