By the Domainex Synthesis Group (Alicia Galván Álvarez, Hugh Tawell, Tenin Traore, Andrew Jones, Jonas Calleja, David Gibson, Fergus Preston, Andrea Bombana, James Sharpe and Venu Komanduri)
In this latest edition of our blog series, we have focused on the following two recent publications, one describing an unusual method of forming sp3-sp2 carbon-carbon bonds and the other forming β,γ-unsaturated ketones from alkenes and carboxylic acids.
- Formal Cross-Coupling of Amines and Carboxylic Acids to Form sp3−sp2 Carbon−Carbon Bonds. Douthwaite et al, JACS, 145, 10930-10937, 2023.
- Direct allylic acylation via cross-coupling involving cooperative N‑heterocyclic carbene, hydrogen atom transfer, and photoredox catalysis. X. Wang et al, Nat. Comm. (2023).
Formal Cross-Coupling of Amines and Carboxylic Acids to Form sp3−sp2 Carbon−Carbon Bonds
The sp3-sp2 carbon-carbon bond is the sixth most prevalent substructure in Drugbank molecules and methods for forming these bonds are expanding. The most common procedures for producing these motifs involve the reaction of an organometallic reagent (organozinc, organomagnesium, organolithium etc.) and a halide, either through nucleophilic substitution or metal-catalysed cross-coupling. Reductive cross-couplings of two halides under nickel catalysis has also been reported which removes the need for pre-functionalisation which minimises potential risks with the stability of organometallic reagents. Although sp3 halides are widely available, they represent an orthogonal region of chemical space to commercially available nucleophiles such as amines. In a similar way, commercially available sp2 halides and pseudo-halides share little homology with sp2 carboxylic acids and a transformation which could realise carbon-carbon bond formation using these coupling partners would allow greater access to these important structural motifs.
To this end, Douthwaite and co-workers1 reported the reductive cross-coupling of N-acyl glutarimides (derived from carboxylic acids) and Katritzky salts (derived from amines) to form sp3-sp2 carbon-carbon bonds using nickel catalysis. While amide bonds are strong due to the π-donation from the nitrogen, N-acyl glutarimides do not sit in a planar conformation and little or no π-donation can occur. This weakened C-N bond is susceptible to oxidative addition by a nickel (0) catalyst to give a nickel (II)-acyl complex. A decarbonylation step gives access to a nickel-aryl complex. The Katritzky salt (N-alkyl-pyridinium salt) contains a weak C-N bond which can be reduced by a nickel/manganese cycle to release the alkyl radical. The inherent instability of this alkyl radical leads it to form a complex with the nickel-aryl species giving rise to a Ni(III) intermediary species. Following this, reductive elimination of the alkyl and aryl groups form the sp3-sp2 carbon-carbon bond.
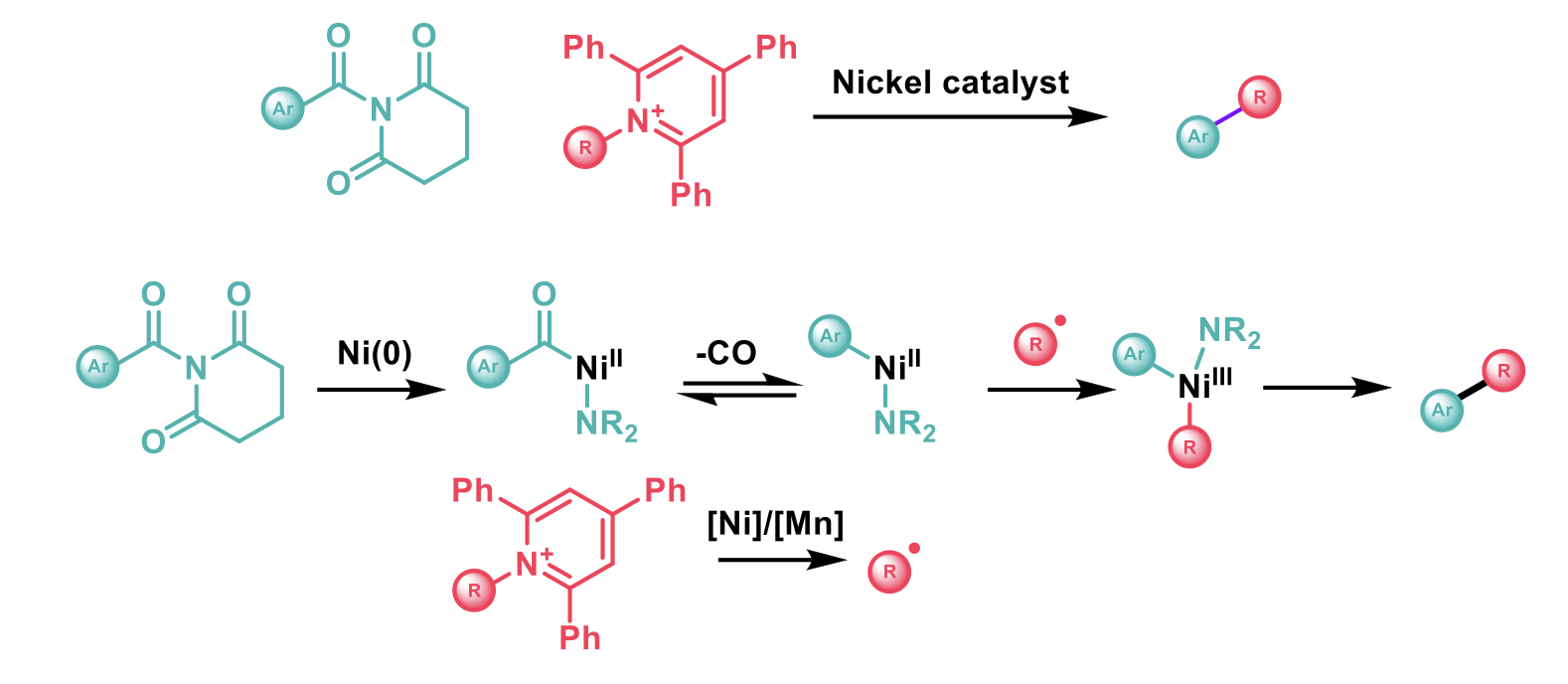
As the generation of N-acyl glutarimides (from benzoic acids) and Katritzky salts (from amines) is relatively straightforward, the authors demonstrated this reaction with a range of substrates with moderate yields. Primary α-primary amines are most suited for this transformation due to the inherent reactivity of primary alkyl radicals and electron-poor aryl groups on the derivatised benzoic acids give higher yields due to both the weaker amide bond and a faster decarbonylation step. Electron-rich acids are poorer substrates as mixtures of the desired product and a ketone product are observed when radical addition to the nickel complex occurs before decarbonylation can take place.
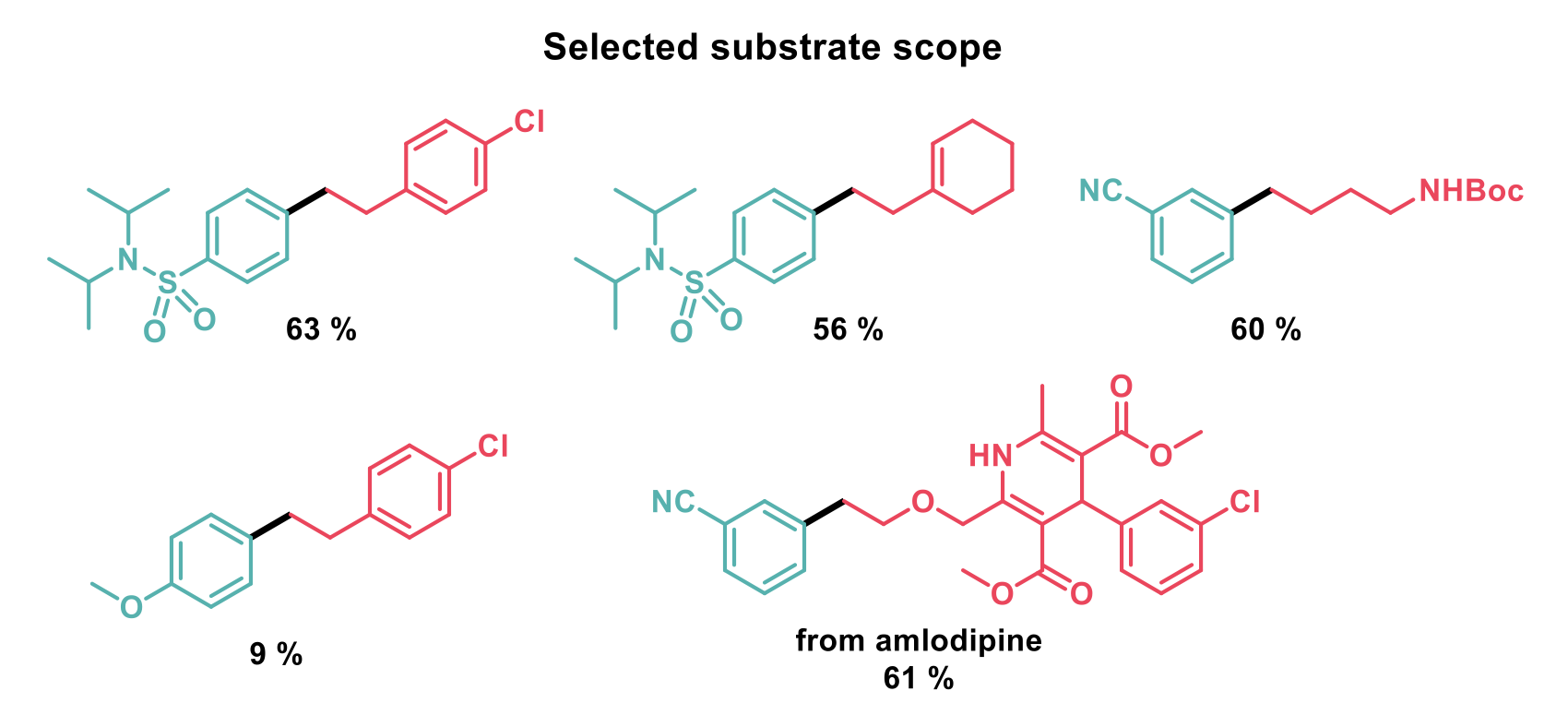
Addition of ruthenium (III) chloride to the reaction sequesters carbon monoxide and promotes the decarbonylation step to give impressive changes in selectivity for naphthalene and acrylic acid derived substrates.

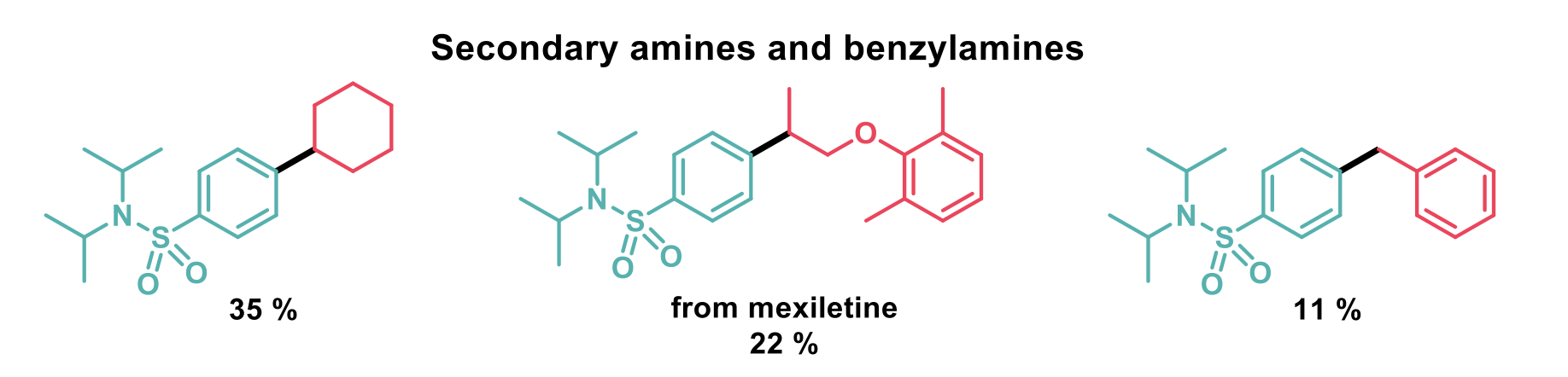
With the initial set of conditions, secondary alkyl pyridinium salts performed poorly due to their higher reactivity towards reductive cleavage. The secondary radicals produced then reacted with their parent pyridinium salts to give dihydropyridine products before reaction with the nickel complex could occur. To alleviate this problem, changing to a more electron-deficient ligand, reducing the temperature and addition of gallium (III) trichloride increased the yields to synthetically useful amounts. As benzylpyridinium salts decompose faster still, only 11 % of product was observed in this case.
Direct allylic acylation via cross-coupling involving cooperative N‑heterocyclic carbene, hydrogen atom transfer, and photoredox catalysis
While the synthesis of α,β-unsaturated ketones is widely known with many orthogonal methods, the construction of β,γ-unsaturated ketones remains challenging. This is despite the prevalence of such structures in bioactive molecules, particularly polyketide natural products. Current methods to form β,γ-unsaturated ketones rely on prefunctionalisation of an alkene as the vinyl bromide, iodonium, zirconate or boronate and reaction with an enolate equivalent using metal catalysis.
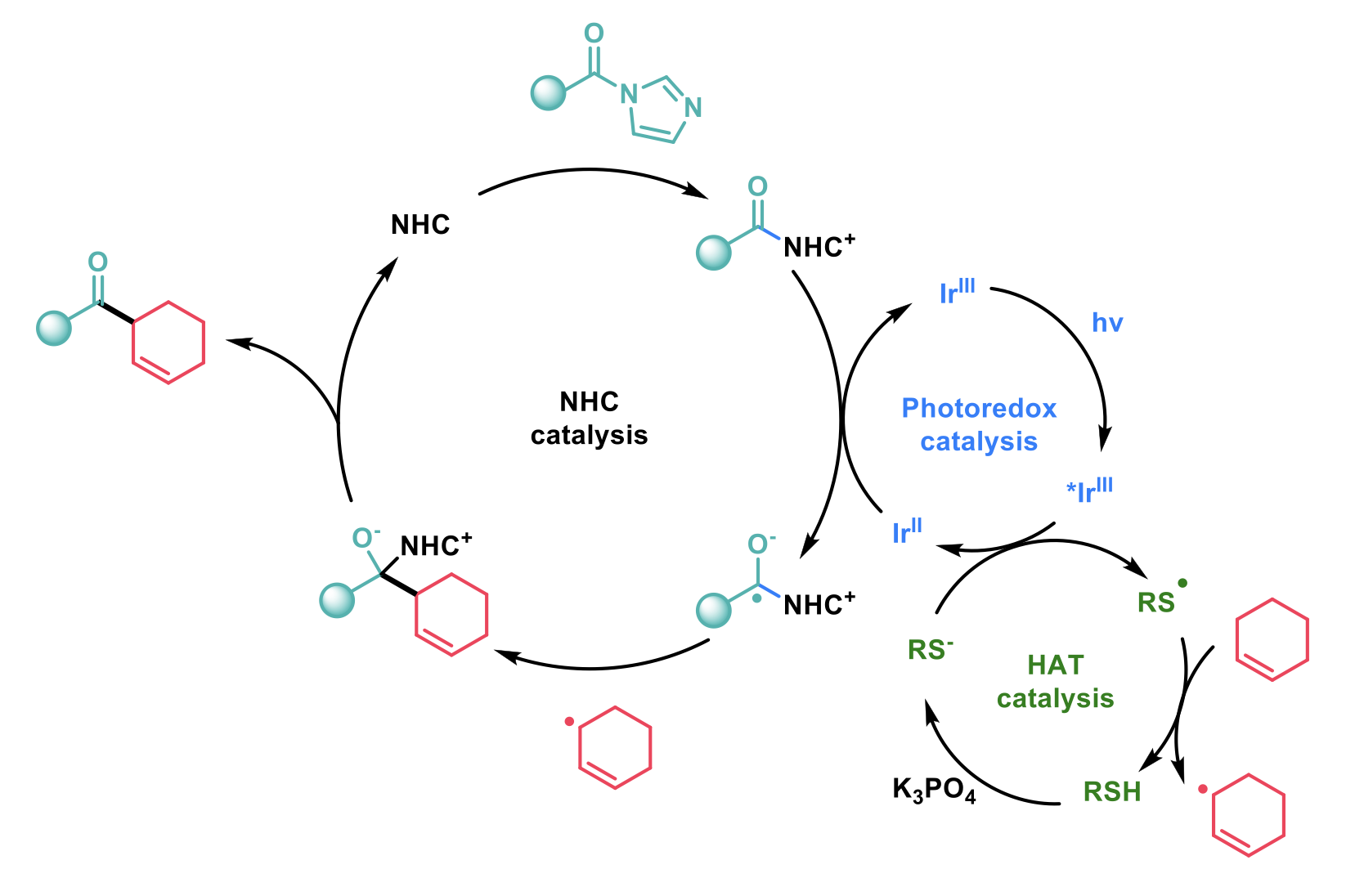
N-heterocyclic carbene (NHC) catalysts have long been employed to enable Umpolung reactivity of carbonyl compounds (e.g. benzoin and Stetter reactions) and the demonstration of these catalysts with radical reactions is growing. Wang and co-workers2 have reported a tricatalytic system marrying NHC catalysis, photocatalysis and hydrogen atom transfer (HAT) catalysis to produce β,γ-unsaturated ketones in a single step from benzoic acid derivatives and feedstock alkenes.

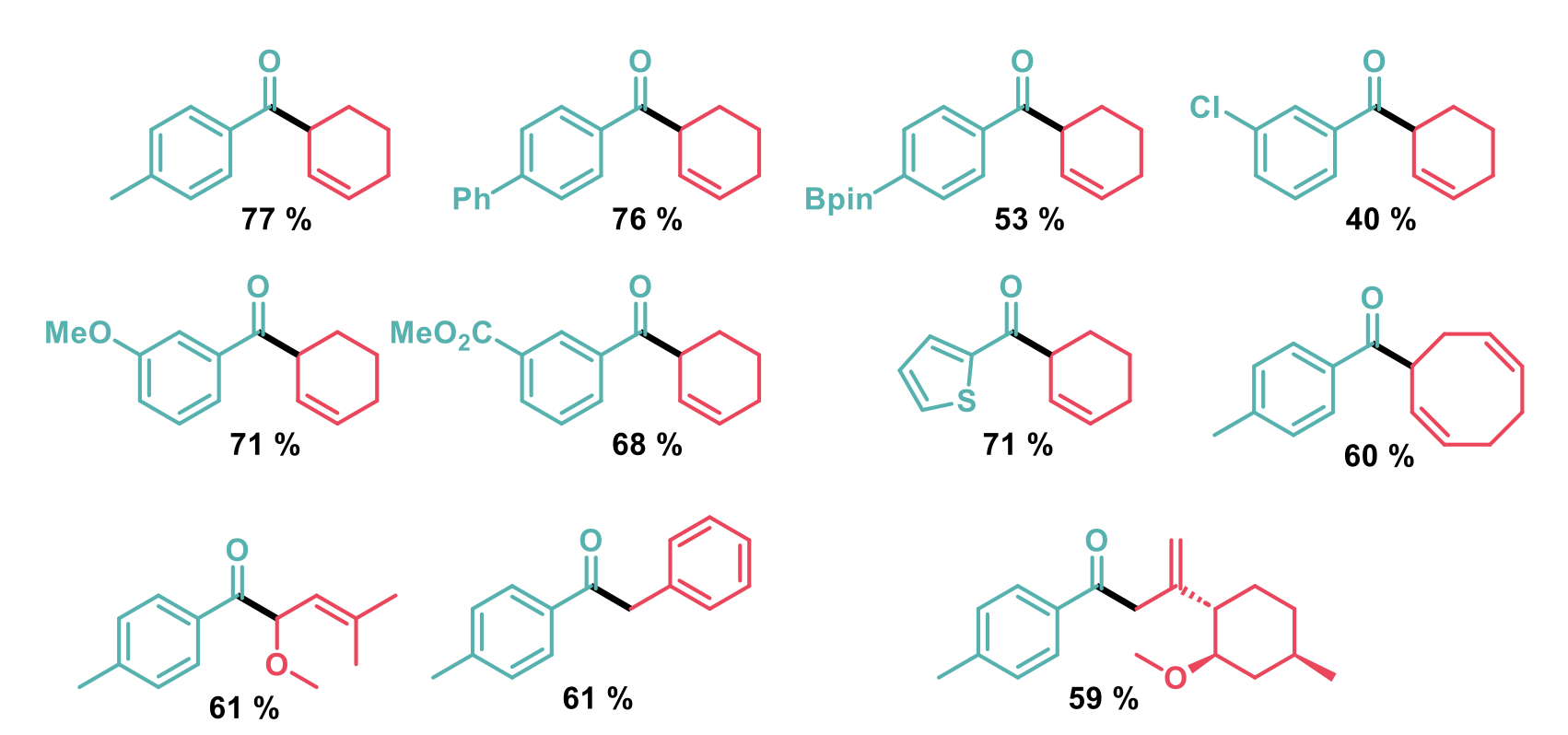
The scope of the reaction was demonstrated to include meta and para-substituted arenes with electron-rich and electron-deficient substituents tolerated. A free phenol was tolerated in the ortho-position and heterocycles also performed well. With respect to the alkene partner, different substitution patterns around the alkene were tolerated, with the selectivity determined by the hydrogen-atom transfer step. Benzylic substrates could also be used as reacting partners to produce 1,2-diaryl ketones in good yield and a limited range of protected oxygen functionality was also tolerated.
The combination of three different types of catalysis requires careful control and balance of the rates of each catalytic cycle. The thiol HAT catalyst is deprotonated by a phosphate base and oxidised by the iridium-based photocatalyst to give a thiol radical which can abstract a hydrogen atom in the α position to an alkene to give an allyl radical. The carboxylic acid, activated as the acyl imidazole, reacts with the NHC catalyst to give an intermediate which can be reduced by the iridium-based photocatalyst to give a ketyl radical. This ketyl radical undergoes radical-radical coupling with the allyl radical to give the β,γ-unsaturated ketone and regenerate the NHC catalyst.
We hope you found this blog interesting. Our next review will be available soon, but in the meantime, get in touch to find out how we can help solve your synthetic chemistry challenges.