By the Domainex Synthesis Group (Alicia Galván Álvarez, Claire Sear, Hugh Tawell, John May, Tenin Traore and Andrew Jones)
In the latest edition of our blog series, we have focused on the following two recent publications, which describe highly useful synthetic transformations:
- Nontraditional Fragment Couplings of Alcohols and Carboxylic Acids: C(sp3 )−C(sp3 ) Cross-Coupling via Radical Sorting. Holt A. Sakai and David W. C. MacMillan. JACS, 2022, 144, 14, 6185–6192.
- Phosphine-Catalysed Enantioselective (3+2) Annulation of Vinylcyclopropanes with Imines for the Synthesis of Chiral Pyrrolidines. Fuhao Zhang, Xuan Dai, Lei Dai, Wai-Lun-Chan, Xiaodong Tang, Xumu Zhang, and Yixin Lu, ACIE, 2022, ASAP.
Radical sorting allows the non-traditional coupling of alcohols and carboxylic acids
Alcohols and carboxylic acids are highly abundant, versatile and operationally convenient organic fragments. These motifs are commonly coupled via the esterification reaction, reported by Fischer and Speier over 125 years ago. However, the direct coupling of alcohols and carboxylic acids to create new C(sp3) – C(sp3) bonds has been an appealing but elusive goal for many years. In the first of our selected publications, David W. C. MacMillan (Nobel prize in Chemistry 2021 jointly with Benjamin List for their development of asymmetric organocatalysis) and co-workers, describe the development of a cross-coupling reaction which achieves this goal.1
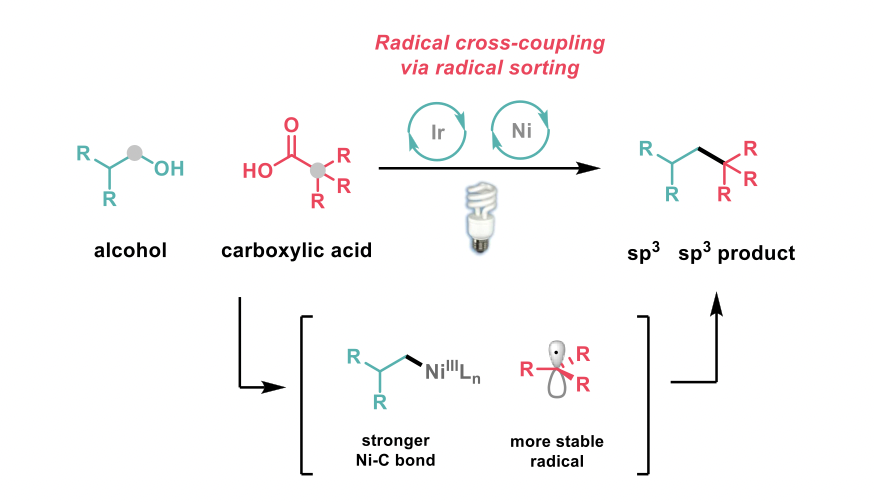
The authors combined an N-heterocyclic carbene (NHC)-mediated deoxygenation of alcohols with a hypervalent iodine-mediated decarboxylation of carboxylic acids, in a dual nickel/photoredox-catalysed technology to prepare a wide array of alkyl-alkyl cross coupled compounds. The main challenge for this non-traditional fragment coupling is ensuring the cross-compatibility of the activation modes and an efficient cross-coupling reaction of the two alkyl radicals generated. In this regard, the authors hypothesised that the nickel catalyst would bind and stabilise the less-substituted alkyl species preferentially promoting the build-up of the more-substituted radical in solution, leading to a “radical sorting” mechanism that would favour the desired species.
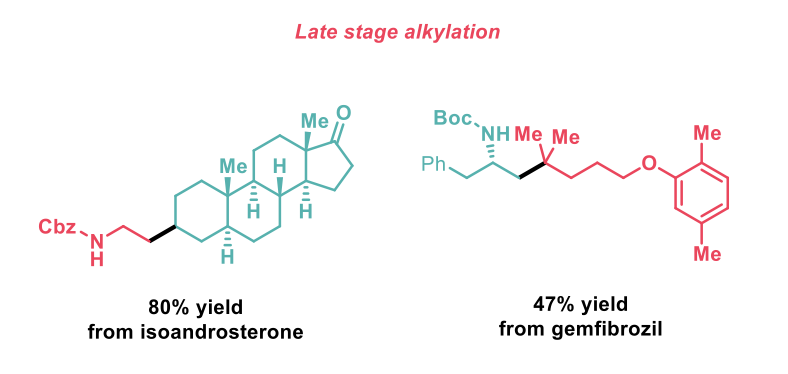
The scope of the reaction includes the formation of challenging quaternary centres and has also been expanded to the C1-deoxyalkylation of alcohols as well as alcohol homologation. Complex alcohol and acid substrates were coupled in useful yields demonstrating the application of this methodology in the context of late-stage functionalisation of drugs, natural products and biomolecules.
Chiral pyrrolidines via metal-free phosphine catalysts from vinylcyclopropanes
The ring-opening of cyclopropanes, driven by the release of ring strain, is a transformation well-utilised in synthetic chemistry, although catalysts are often required to facilitate the process due to the kinetic stability of the substrates. In the second publication we selected, Yixin Lu et al2 describe the development of an asymmetric ring opening using chiral phosphine catalysts to give chiral pyrrolidines, which are key structures in numerous biologically relevant molecules. The cyclopropane ring opens up to give a zwitterionic intermediate, which can then undergo a (3+2) annulation with an imine, giving rise to the pyrrolidine product.
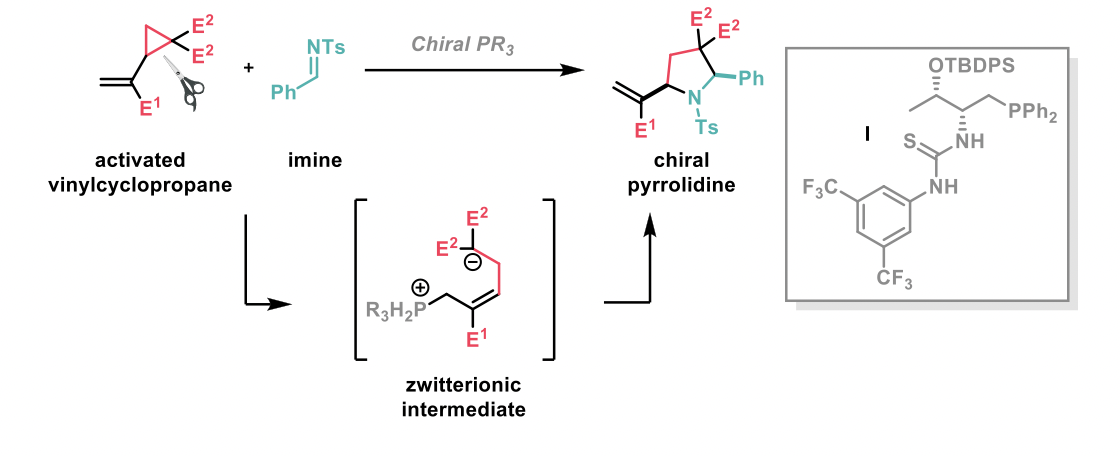
An extensive optimisation by the authors gave rise to catalyst I as the best chiral phosphine for this transformation, resulting in very good chiral selectivity (95% enantiomeric excess) for the trial substrates. It was observed that the catalyst could also react with the annulation product resulting in loss of ee, however this was rectified by the authors by including an excess of the cyclopropane starting material. The scope of N-tosylimines was explored and extended to a number of electron-withdrawing and donating moieties, heterocycles and vinylic substituents. Aliphatic imines were also well-tolerated, albeit with lower enantiomeric excess for those less sterically hindered. Different ester groups on the cyclopropane ring could be utilised as the electron-withdrawing group, including Me, Et, and CF3, as well as a nitro group in place of the ester.
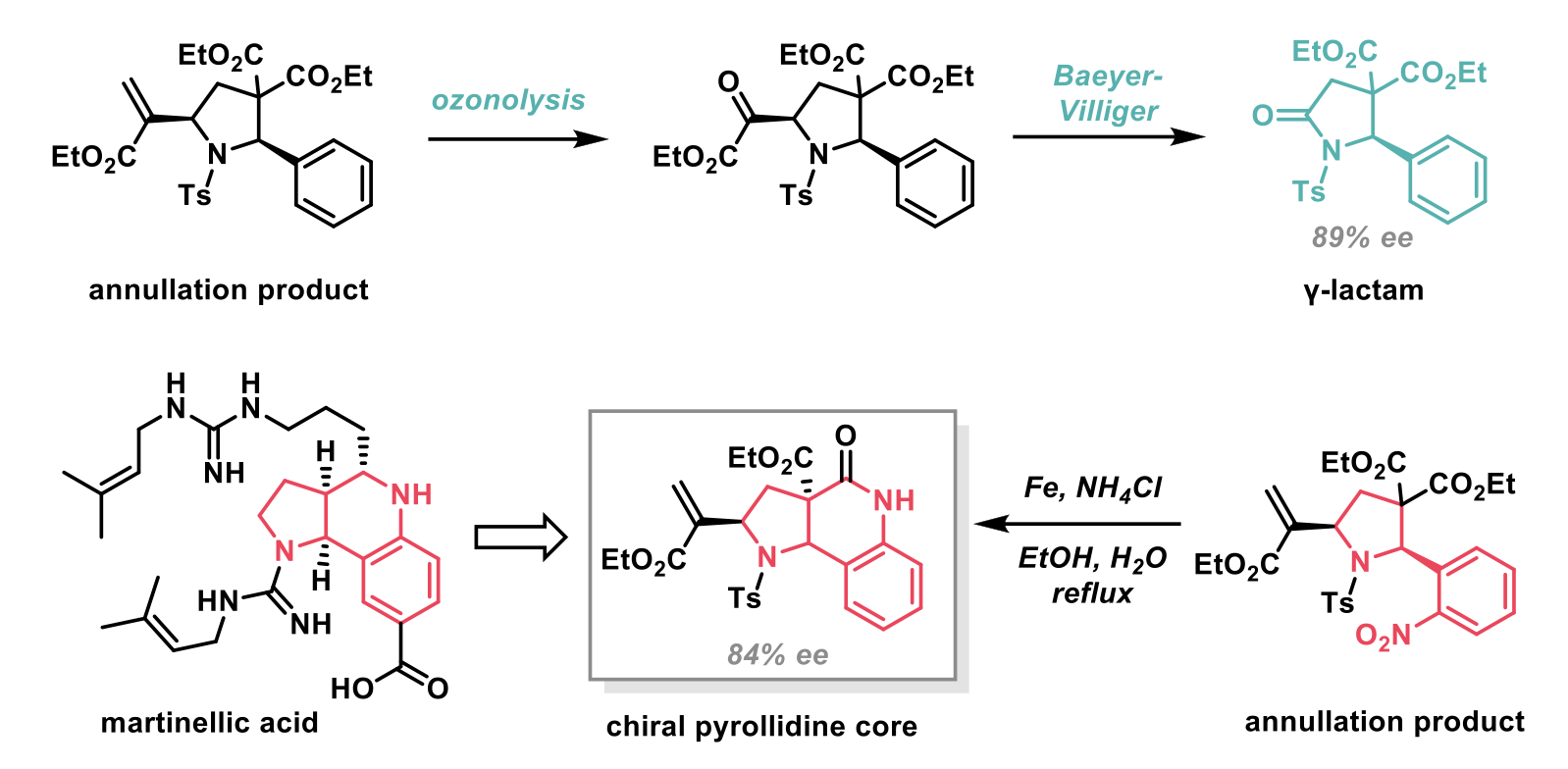
The authors demonstrated the synthetic utility of this methodology by synthesising a series of biologically relevant molecules, including a γ-lactam, via ozonolysis and subsequent Baeyer-Villiger reaction of the pyrrolidine product, and through the synthesis of the pyrrolidine core of martinellic acid (a naturally occurring bradykinin receptor antagonist).
We hope you found this blog interesting. Our next review will be available soon, but in the meantime, get in touch to find out how we can help solve your drug discovery challenges.