This article was originally published in MDC Connects: A Guide to Drug Discovery which can be downloaded here.
The chemistry discipline is instrumental at all stages of drug discovery, from the creation of diverse or targeted libraries for screening, through design, synthesis, formulation and ultimately the scale up and manufacture of the final drug product.
In the MDC Connects webinar we heard from three chemists. Trevor Perrior described how fragment-based drug design can be applied, taking us through a case study which illustrates how low molecular weight fragment hits can be turned into drug candidates. James Hitchin focussed on synthetic chemistry and the different skills required from early Drug Discovery to Development phases, with particular reference to the way in which transformation maps can be applied to guide decision making. Alison Foster described how the formulation of a compound can be optimised to maximise dose and absorption in preclinical studies and enhance the properties of the API to meet the Target Product Profile in the clinic.
View the recording and slides for the webinar.
IKKɛ/TBK1: A case study of approaches to turning fragment hits into drug candidates
Domainex is a leading provider of high quality innovative and efficient scientific solutions that enable successful drug discovery programmes against a wide range of drug targets. Amongst its clients are pharmaceutical, biotechnology, academic institutions, and patient foundations from all around the world.
Fragment-Based Drug Design (FBDD) is an approach where libraries of low molecular weight compounds are screened to identify highly-efficient chemical starting points. Skilful elaboration of these very efficient fragment hits with the addition of molecular weight only where this is of optimal benefit affords potent and bioavailable drug candidates.
This case study illustrates the use of FBDD to invent an efficient and selective drug candidate against IKKɛ and TBK1. Both of these kinases play an important role in regulating the immune response in a group of inflammatory diseases known as interferonopathies e.g. systemic lupus erythematosus, Sjogren’s syndrome, and scleroderma. This class of autoimmune diseases are caused by upregulation of interferon signalling leading to inappropriate activation of interferon-stimulated genes, many of which lead to the expression of inflammatory mediators. Inhibition of IKKɛ and TBK1 will prevent this upregulation and reduce the pro-inflammatory signals. The development of small-molecule inhibitors has been challenging, and the biologic treatments that are available for interferonopathies are often ineffective and expensive. Therefore, there is a very high unmet medical need for specific and effective treatments for these diseases.
Key results
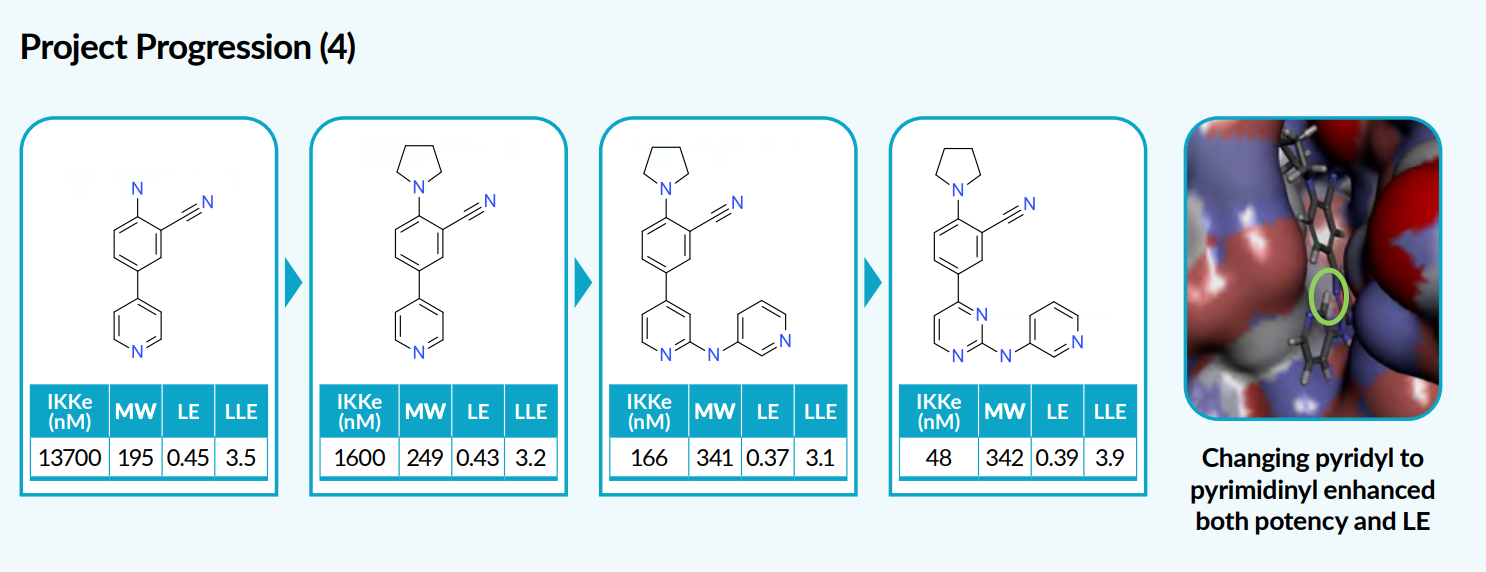
By screening a fragment library, a hit was selected based on its potency against the target, molecular weight, and a ligand-efficiency metric. Enzyme crystal structure studies revealed opportunities to increase potency without affecting efficiency. By manipulating the initial hit fragment by the substitution of an amine, followed by aryl or heteroaryl amino substitution on the pyridyl ring, the potency of the compound was increased with no loss in ligand efficiency. A final replacement of a pyridine with a pyrimidine ring enhanced potency further to give a 50 nM compound with a distinctive cyano group.
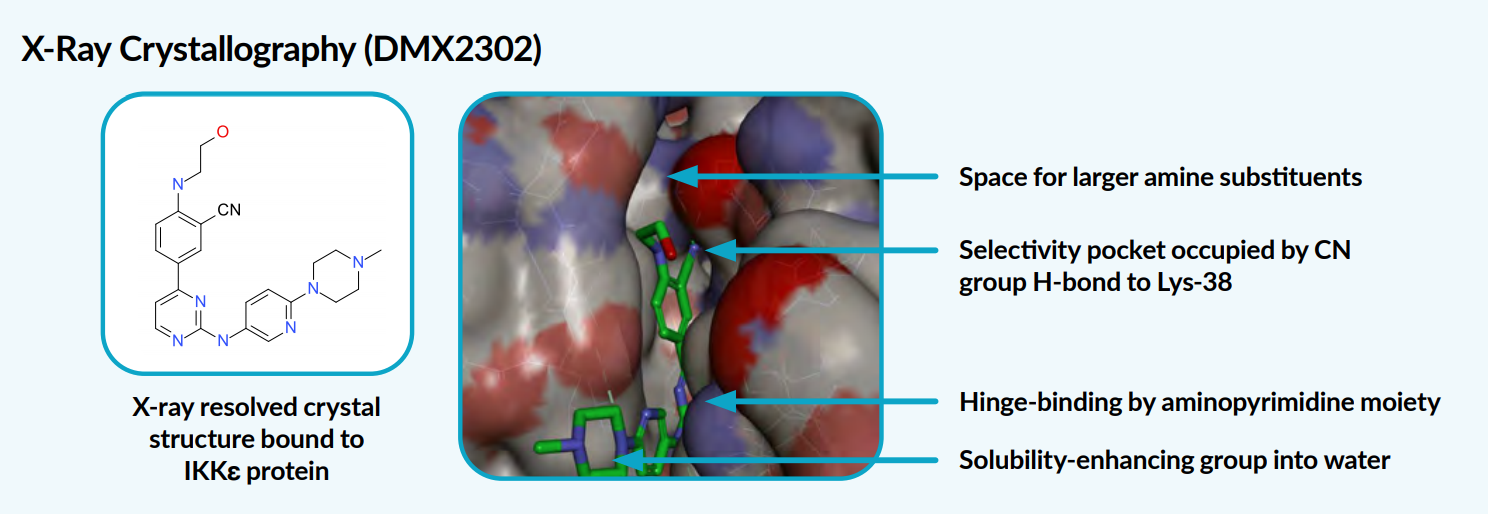
X-ray crystallography provided a high-resolution crystal structure of an exemplar protein-ligand complex, where the CN group fitted into the narrow pocket in the kinase, forming a hydrogen bond to lysine 38.
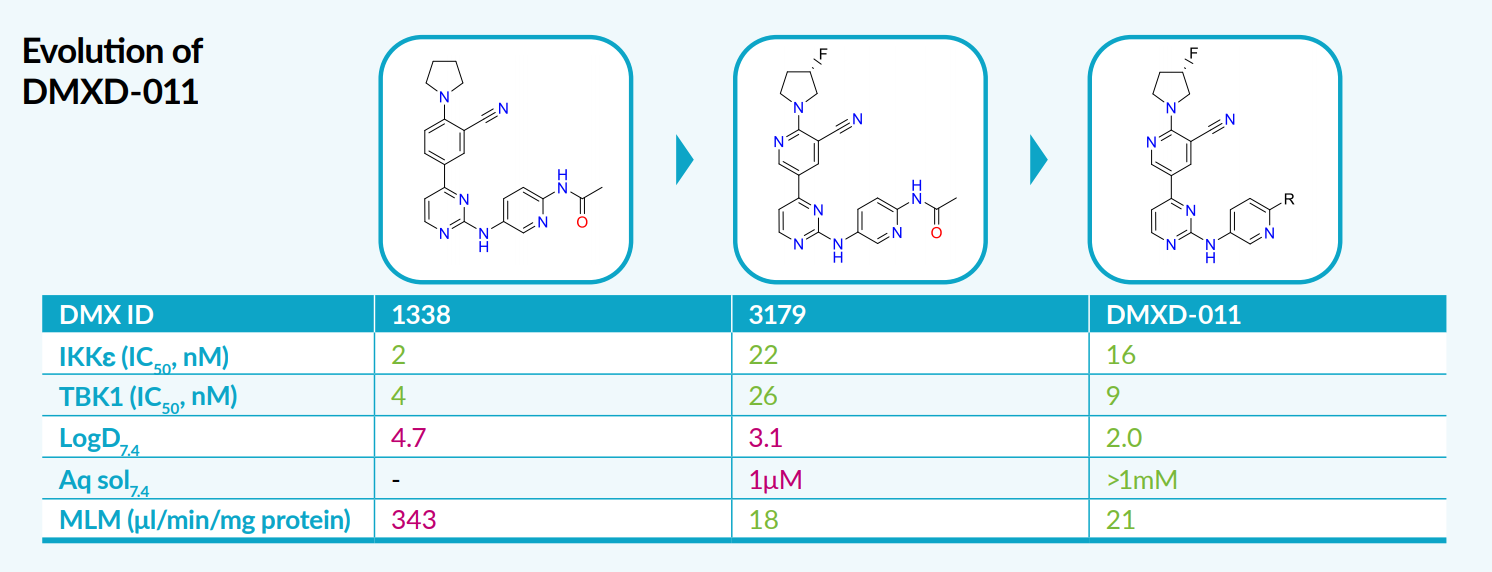
Structure-based drug design and manipulation of molecular and physical properties allowed the solubility, metabolic stability and permeability to be further enhanced. By lowering the logD with the introduction of polar atoms and substituents, improved metabolic stability was achieved whilst maintaining the high selectivity and potency and DMXD-011 was identified.
Will this work in patients?
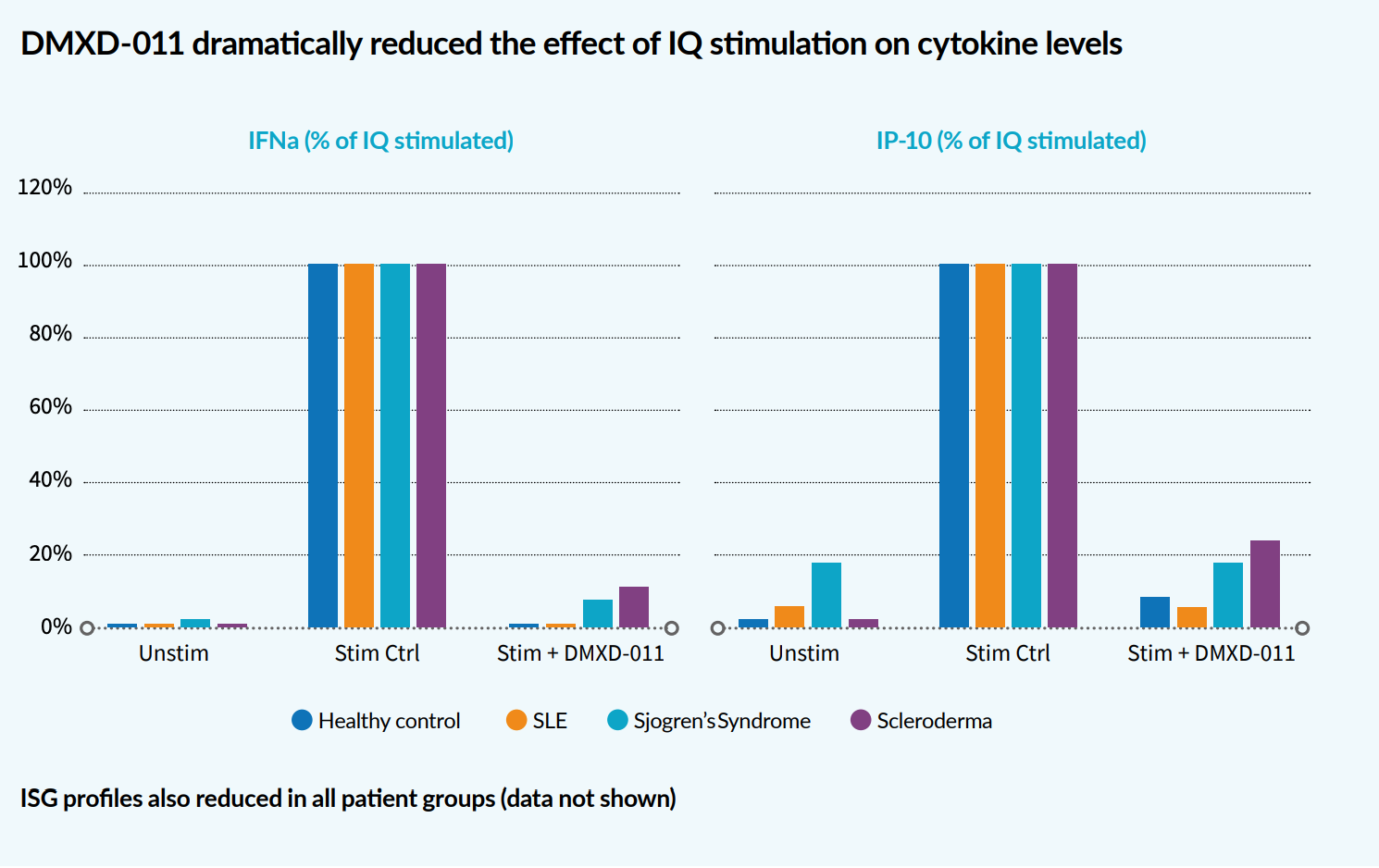
Ex vivo studies using inflammatory cells from healthy donors and patients with interferonopathies showed that the compound almost completely blocked the release of interferon-alpha and other inflammatory mediators, leading to reduced expression of interferon-stimulated genes demonstrating excellent proof of concept.
How we did it
These results were achieved using a FBDD approach based on identifying small chemical fragments which bind to the biological target. Here Domainex fragment libraries were screened using a high concentration biochemical assay against the IKKɛ enzyme, and a unique in-house crystal structure of IKKɛ was obtained using a domain approach. Through structure-based drug design and profiling with a cascade of enzyme and cellular assays, we optimised the compound. By using the FBDD strategy we efficiently designed DMXD-011, a unique low clearance, metabolically-stable IKKɛ/TBK1 inhibitor, that was validated in proof-of-concept studies in animal models and an ex vivo human assay. DMXD-011 is now in pre-clinical development.
Download MDC Connects: A Guide to Drug Discovery.
About the Author - Trevor Perrior
Trevor is now a technical consultant to Domainex. Following his education at the University of Cambridge Trevor undertook academic research at Cambridge, Oxford and in the USA. He then held a number of senior R&D leadership roles at ICI, Zeneca, AstraZeneca and Celltech, where he worked in the UK, USA, and Switzerland. In 2005 Trevor joined NCE Discovery as Chief Scientific Officer, and when NCE Discovery merged with Domainex he became Director of Research for the enlarged company. In 2016 he was appointed Chief Scientific Officer of Domainex, and then in 2018 Chief Executive Officer. In April 2020, Trevor retired from this role, but continues to support Domainex as a scientific advisor. Trevor is also a scientific consultant to a number of venture capital and charitable funds.