High throughput screening (HTS), the testing of large numbers (from several hundred thousand up to a few million) of diverse small molecules (of molecular weight 400-650 Da for the most part) has been a mainstay of hit identification for drug discovery since the early 1990s. Over this time there have been many advances in HTS – increasing scale and quality of the libraries, significant miniaturisation of assays, advancement of detection technologies and establishment of automation – that ensure diversity-based screening remains the preferred approach to hit finding in many instances.
The downside of HTS is the significant investment required in building the infrastructure to support an efficient HTS operation. Furthermore, there remains significant difference of opinion on the successfulness of the approach in identifying suitable hit matter; with typical hit rates of ~1%, there is significant inherent redundancy in the process. Some sources report successful outcomes from screens in only 50% of cases, whereas others believe it to be much higher(1,2). There is even more debate on the overall impact that HTS has on development pipelines(3). It is also a matter of opinion whether the compound libraries that have been built and curated by many organisations, over many years, remain a valuable source of hit matter based on our current understanding of drug-like properties, and indeed what size should that library be(4). This is due to the shift away from classical approaches such as G-protein coupled receptor antagonism and ATP competitive kinase inhibition towards other target classes such as transporters, protein-protein interactions and enzymes that exert their effect on DNA plus the pursuit of allosteric and covalent modes of action.
The uncertainty of success and the time and costs of screening large numbers of compounds are significant concerns, particularly for smaller organisations. Fortunately, advances in computer aided drug design (CADD) and protein crystallographic techniques have opened up new approaches which are now equally mainstream for hit identification. Virtual screening, similar to HTS, relies upon assessing a large, diverse library of compounds. The difference is that the compounds in a virtual screening library are contained in databases rather than being physically assembled by the organisation conducting the screen. Using knowledge of the target protein(s) based on a crystal structure(s), homology models or a predicted protein structure and knowledge of ligands that may bind to the target, large numbers of molecules (typically over 1 million) are screened computationally in a matter of days. This is based on 2D and 3D shape and charge for docking into the assumed binding pocket(s) of interest.
Having completed the computational screen, a prioritised list of compounds, effectively a targeted set (usually less than 1,000) are sourced (typically from commercial vendors) for physical screening against the target protein of interest. This can often result in enriched hit rates of up to 5% with potencies in the single-double digit micromole range, compared to HTS hit rates which are typically ~1%. As virtual compounds can be pre-filtered based on their drug-like properties, the resultant hits can be fast-tracked into down-stream medicinal chemistry to further optimise these properties. The knowledge gained can also inform further refinement of the computational model to allow additional rounds of virtual screening and an updated selection of compounds to be made.
It would be wrong however, not to acknowledge the potential downsides of virtual screening, of which the main one is serendipity. In a conventional HTS, no assumption is made of the types of molecules being sought and the approach is agnostic of the binding site one is wishing to exploit. This is not the case for virtual screening where some definition of initial parameters is inevitable. This may become less of an issue as artificial intelligence (AI) is increasing deployed in drug discovery. However, it is too early to determine if the promise of this approach will be realised and whether AI will take over from HTS as the mainstay of hit finding and the navigation of the complex process that is drug discovery(5).
Until this point, we have focussed on the screening, either physically or virtually, of small molecules as the source of hit molecules. By and large these libraries are built around industry knowledge on the properties of drug-like molecules and the so-called Rule-of-5(6). The challenge is that the structures of many compounds within a screening library are far from optimal for the binding pockets of new targets, leading to low potency and low efficacy in screening assays. The answer, therefore, lies in reducing the size or fragmenting the small molecules into their core components to aid access to key binding sites, so called fragment-based drug discovery (FBDD). Whilst the use of fragments isn’t a new concept, having been part of drug discovery for approximately 25 years(7), it arguably has not quite attracted the attention afforded to HTS.
Fragment libraries contain far fewer test molecules than a HTS library (usually only 1,000-3,000 entities), the compounds are much smaller (typically with a MW <300 Da) and, analogous to drug-like small molecules, they typically follow a Rule-of-3(8) (see Table 1). Domainex’s library of >1000 fragments generally adheres to the Rule-of-3 criteria (see Figure 1). The downside of reducing the molecule size is that compound affinity and efficacy reduces. Fragment screening is therefore best undertaken using biophysical methods to detect direct binding to the target protein e.g. Surface Plasmon Resonance (SPR)/Grating Coupled Interferometry (GCI) or similar techniques such as MicroScale Thermophoresis (MST) or Differential Scanning Fluorimetry (DSF). Key though for FBDD is to have the crystal structure of the protein of interest. Having identified the fragments that bind to the target protein, crystallographic methods allow the nature of the interaction between the binding site and the fragment to be understood. This allows medicinal chemists to then develop a strategy to grow the fragment in suitable vectors to improve both affinity and efficacy to give an optimised small molecule for the target of interest.

Table 1: Comparison of the Rule-of-5 and Rule-of-3
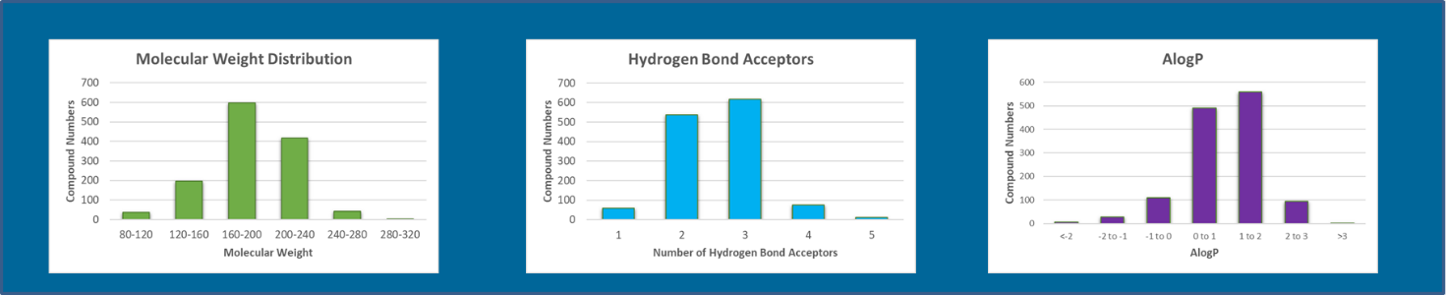
Figure 1: Some molecular and physiochemical properties of the Domainex Fragment Library, showing how the fragments generally adhere to the Rule-of-3 criteria.
The overriding question regarding these three approaches – HTS, virtual screening and FBDD – is: which is the correct strategy to follow? Ultimately, this is target dependent and often driven by the historical success of a particular approach within an organisation. Overall, the timeline to complete the virtual screen is usually shorter than an HTS for a similar size compound library. FBDD again relies upon the initial screening of a small compound set and because of the use of biophysical screening methods, it is known from the outset that you have target engagement. This is unlike HTS, where there is potential for off-target activity due to interference with the assay system. Moreover, the hit finding cost in terms of protein requirements, assay reagents and laboratory consumables are far lower for virtual screening and FBDD compared to HTS. Arguably, the former two approaches enable faster entry into the hit-to-lead and lead-optimisation stages of the drug discovery cascade(9,10).
References
- Fox, S., Farr-Jones, S., Sopchak, L. High-Throughput Screening: Update on Practices and Success. J. Biomol. Screen. 2006, 11, 864–869.
- MacArron, R., Banks, M.N., Bojanic, D. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov. 2011, 10, 188–195.
- Swinney, D., Anthony, J. How were new medicines discovered? Nat Rev Drug Discov. 2011, 10, 507–519.
- Volochnyuk, D.M., Ryabukin, S.V., Moroz, Y.S. et al. Evolution of commercially available compounds for HTS. Drug Discov. Today. 2019, 24(2), 390-402.
- Kim H., Kim E., Lee I., Bae B., Park M., Nam H. Artificial Intelligence in Drug Discovery: A Comprehensive Review of Data-driven and Machine Learning Approaches. Biotechnol Bioprocess Eng. 2020, 25(6), 895-930.
- Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997, 23(1-3), 3-25.
- Erlanson, D., Fesik, S., Hubbard, R. et al. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016, 15, 605–619.
- Congreve M., Carr R., Murray C., Jhoti H. A 'rule of three' for fragment-based lead discovery? Drug Discov Today. 2003, 8(19), 876-7.
- Chen, H., Zhou, X., Wang, A., et al Evolutions in fragment-based drug design: the deconstruction–reconstruction approach. Drug Discov. Today. 2015, 20(1), 105-113.
- Vázquez J., López M., Gibert E., et al. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules. 2020 25(20), 4723.