By the Domainex Synthesis in Review Group (Alicia Galván Álvarez, Claire Sear, Hugh Tawell, John May, Jonás Calleja Priede and Tenin Traore)
At Domainex we regularly review the literature to make sure we are aware of the latest developments. Therefore, we thought we would start a blog series highlighting exciting new synthetic chemistry articles. In this first blog, we have selected two recently published papers to focus on.
- Chemoselective, Scalable Nickel-Electrocatalytic O-Arylation of Alcohols. Hai-Jun Zhang, Longrui Chen, Martins S. Oderinde, Jacob T. Edwards, Yu Kawamata and Phil S. Baran; Angewandte Chemie International Edition, 2021, 60, 38, 20700-20705.
- Carbon Atom Insertion into Pyrroles and Indoles Promoted by Chlorodiazirines. Balu D. Dherange, Patrick Q. Kelly, Jordan P. Liles, Matthew S. Sigman, and Mark D. Levin. J. Am. Chem. Soc. 2021, 143, 30, 11337–11344.
Electrochemistry is the new enabler: O-arylation of alcohols

Aryl-alkyl bond formation represents one of the most employed transformations in the pharmaceutical industry. Different tactics that use Pd-, Cu- and Ni-based catalytic systems have been developed over the past years, as an attractive alternative to classic SN2-based reactions. In this recent publication from Phil S. Baran et al1, a new Ni-catalysed electrochemically promoted reaction is described, which uses mild conditions and achieves a broad substrate scope in an operationally simple way.
Interestingly, the authors describe the synthesis of a BET inhibitor intermediate which could not be successfully synthesised through transition metal catalysed methods or by using photochemical conditions, presumably due to the presence of tertiary amines which are ubiquitous motifs in pharmaceutical molecules.
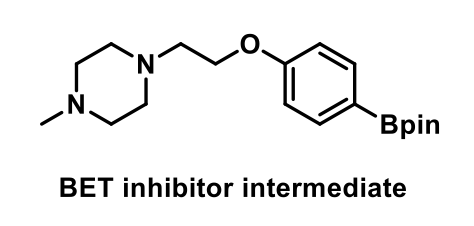
Different studies were developed based on previous electrochemical amination (e-amination) reactions, as well as control experiments that reinforced important aspects of the reaction, such us the necessity of electricity for the reaction to proceed, the crucial role of the Ni-catalyst under basic conditions and the lack of reaction by the use of chemical reductant, such as Zn powder. The method uses mild reaction conditions and does not requite laborious procedures for degassing or any glovebox.
The method can be scaled up either in batch or in continuous flow, delivering 20 g of the corresponding O-arylated products in good yields and, according to the authors this work, presents the widest substrate scope among all the similar methods published so far.
Expand your indole ring!
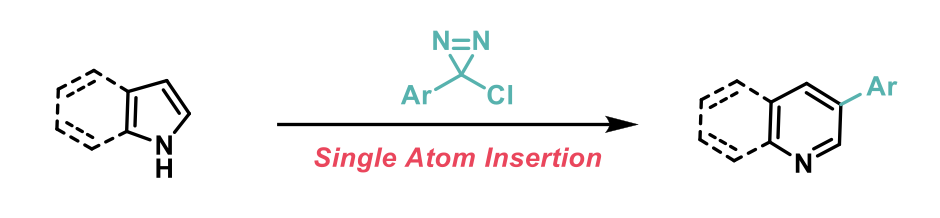
“Single-atom” manipulations represent the paradigm of retrosynthetic simplicity. In this recent JACS report, Levin and co-workers at the University of Chicago2 have taken this to a whole new level developing a new method that selectively generates 3-arylpyridines and quinolines from the corresponding pyrrole and indole counterparts. The reaction proceeds through a C-C insertion of a carbynyl cation, generated “in situ” from dinitrogen extrusion from the corresponding α-halochlorodiaziridines (Chlorodiaziridines are readily prepared in a single-step for the corresponding amidinium salts).
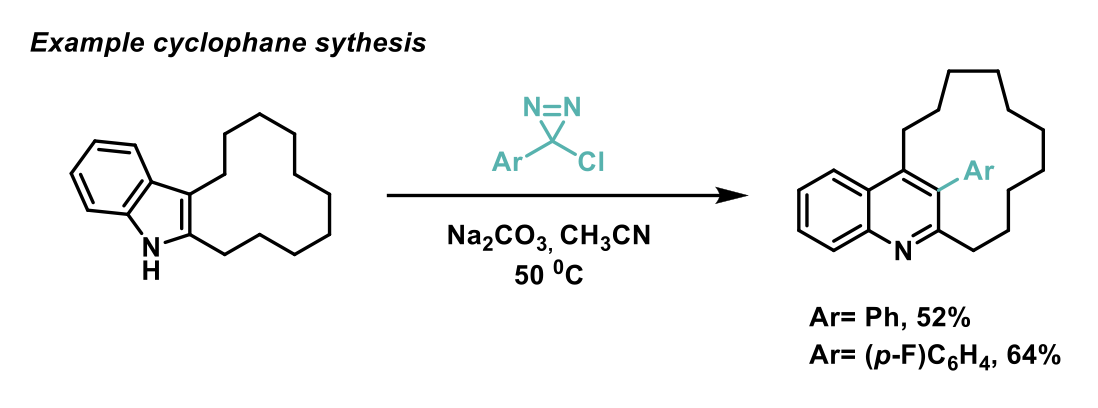
Density Functional Theory (DFT) calculations and mechanism investigations supported the theory that cyclopropanation followed by ring expansion occur during this C-C insertion reaction. The authors found that regioselectivity is controlled by steric effects and they apply that to selectively generate cyclophane derivatives in only two steps, providing straightforward access to a class of compound that are otherwise challenging to synthesise.
The prevalence of pyrrole and indole cores in biologically active molecules means that this method has potential application in the late-stage functionalisation of drugs. This application was demonstrated by the authors by using this method to generate Molindone (Moban) and a Lipitor derivative, thus showing the potential of this methodology to generate molecular complexity in drug discovery programs.
We hope you’ve found this interesting, if so look out for our next synthesis review which will be available on our website soon and get in touch to find out how we can employ the latest synthetic approaches to solve your drug discovery challenges.