By the Domainex Synthesis Group (Andrea Bombana, Hugh Tawell, Andrew Jones, Venu Komanduri, Brahmam Medapi, Robyn Presland, and Vinny Duong)
In this blog post, we review two recent publications that highlight innovative and impactful synthetic methodologies. The first explores skeletal editing of saturated N-heterocycles via N-atom insertion to access previously challenging scaffolds, while the second introduces a traceless directing strategy for selective C7 C–H functionalisation of quinolines using copper catalysis.
- Skeletal Editing of Pyrrolidines by Nitrogen-Atom Insertion. Jinghao Li, Pengcheng Tang, Yang Fan and Hongjian Lu. Science, 2025, 389, 275-281.
- Formal C-H Functionalization of Quinolines at the C7 Position Through a Traceless Directing Strategy. Kun Zhou, Ni Zhang, Xin-Rou Zhong, Rui-Rui Wu, Yang-Jie Mao, Peng Ye, Gen Luo, Dan-Qian Xu and Shao-Jie Lou. Angew. Chem. Int. Ed. 2025, e202514319.
Skeletal Editing of Pyrrolidines by Nitrogen-Atom Insertion
Skeletal editing is becoming a highly valuable approach for the modification of cyclic cores to provide access to a range of scaffolds without the need for targeted construction. Commonly focusing on single atom insertion or deletion, skeletal editing allows for late-stage alteration of cyclic ring sizes. This presents a great opportunity for acceleration of discovery and optimisation of medicinally relevant compounds. In addition, the approach has the potential to grant access to chemical space that was previously perceived as challenging or ‘unattainable’.
Given their importance and relevance, much of the initial efforts for skeletal editing have been focused on nitrogen containing heterocycles. Previous reports demonstrate nitrogen-atom (N-atom) insertion of N-heterocycles with Morandi, Ackermann and Sharma all harnessing nitrene intermediates for the conversion of indoles to quinolines. More recently, Giazza and Zheng have achieved N-atom insertion of pyridines to afford diazepines through photocatalytic strategies. Whilst this previous work shows the applicability towards π-systems, strategies for skeletal editing of saturated systems remain a challenge, in part due to the lack of π-bond activation pathways. However, given the abundance of saturated N-containing heterocycles (pyrrolidines, piperidines, piperazines etc.), strategies for harnessing these for single atom modification are highly desirable.
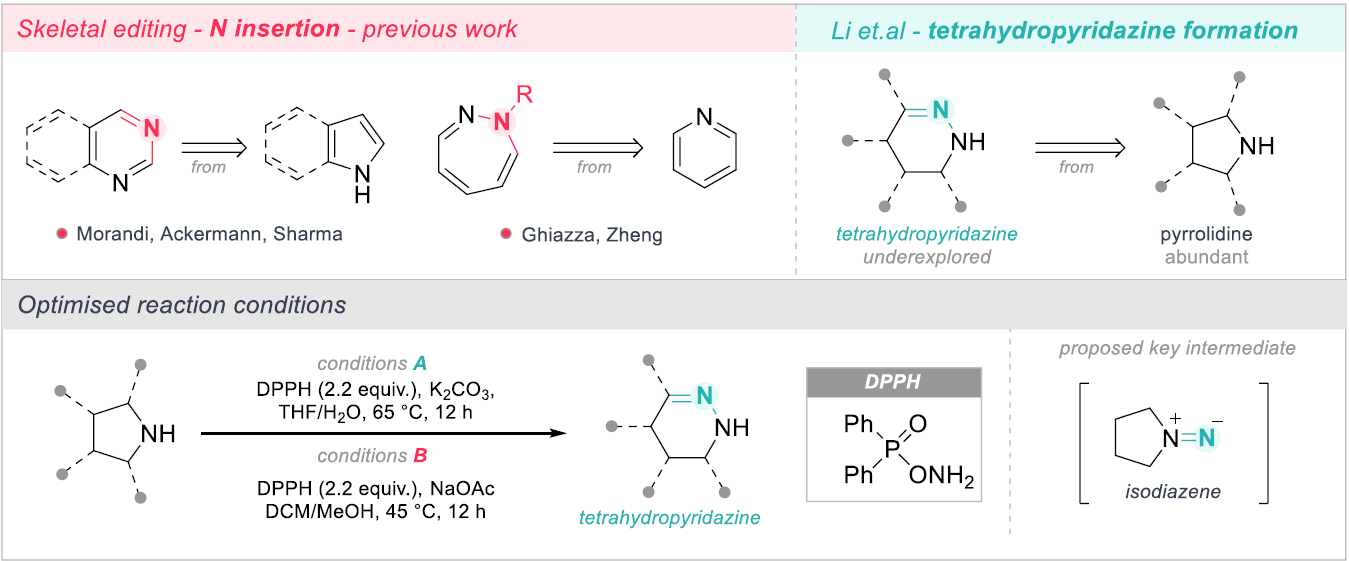
Scheme 1: Transformation of pyrrolidines to tetrahydropyridazines.
To this end, Lu and co-workers have reported the conversion of pyrrolidines to diazacyclic tetrahydropyridazines (Scheme 1). This core has been somewhat underexplored in comparison to structurally related structures such as pyrimidines and piperazines due to its challenging synthesis. The reported procedure makes use of O-diphenylphospanyl hydroxylamine as an electrophilic nitrogen donor, which proved superior compared to alternative hydroxylamine-based electrophiles. Following optimisation, two sets of standard conditions were identified. The first set of conditions include the use K2CO3 as a base, whereas the second set of conditions omit the base completely. Interestingly, the reaction showed great sensitivity to the base use with a large variation observed for K2CO3, KOH, NaOAc, pyridine and NaOMe. Mechanistic studies indicate that the reaction proceeds via an isodiazene intermediate, which undergoes a 1,3-hydrogen shift, ring opening, and rapid cyclisation to yield the N-inserted product.
The scope of the reaction showed applicability to a wide range of substrates, with moderate to excellent yields achieved for tetrahydropyridazine formation from a broad array of pyrrolidines. Interestingly, some substrates required conditions A and some required conditions B for effective N-insertion, highlighting the substrate specific nature of the transformation. α-substituted pyrrolidines typically performed well in the reaction, however β-substituted pyrrolidines presented regioselectivity issues yielding mixture of regioisomers. The introduction of electron-withdrawing groups greatly improved the regioselectivity on these substrates, showing preference for N-insertion at the more electron-deficient C-N bond. The reaction also demonstrates effective skeletal editing beyond monocyclic systems, with fused, spirocyclic and bridge pyrrolidines all showing good conversion. The authors note that most systems tested are readily available pharmaceutically fragments. This is exemplified further by the construction of simple N-functionalized tetrahydropyridazines showing promising biological activity and also through the N-insertion of fragments derived from bioactive analogues (Scheme 2). Overall, given the broad applicability of the method, access to previously perceived challenging scaffolds has been unlocked, paving the way for the exciting development of next-stage pharmaceuticals.
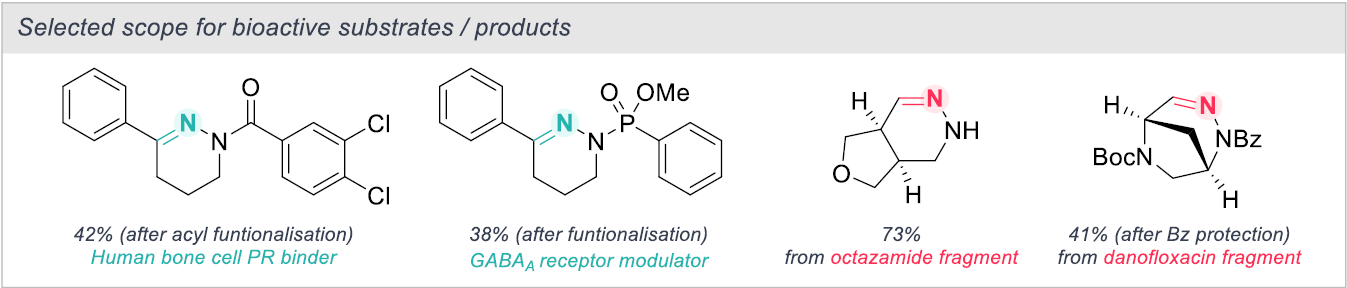
Scheme 2: Substrate scope for bioactive substances/products.
To further highlight the synthetic utility and wider use of the method, the authors demonstrate synthesis of labelled tetrahydropyridines through skeletal editing of Melanostatin (MIF-1) and a fragment of Seltorexant (Scheme 3). Skeletal editing of a piperidine to form a 7-membered diazacycle was also shown to proceed in good yield (61%). The versatility of the tetrahydropyridazine products formed was illustrated by efficient reduction and oxidative aromatisation to the corresponding piperidazine and pyridazine respectively.
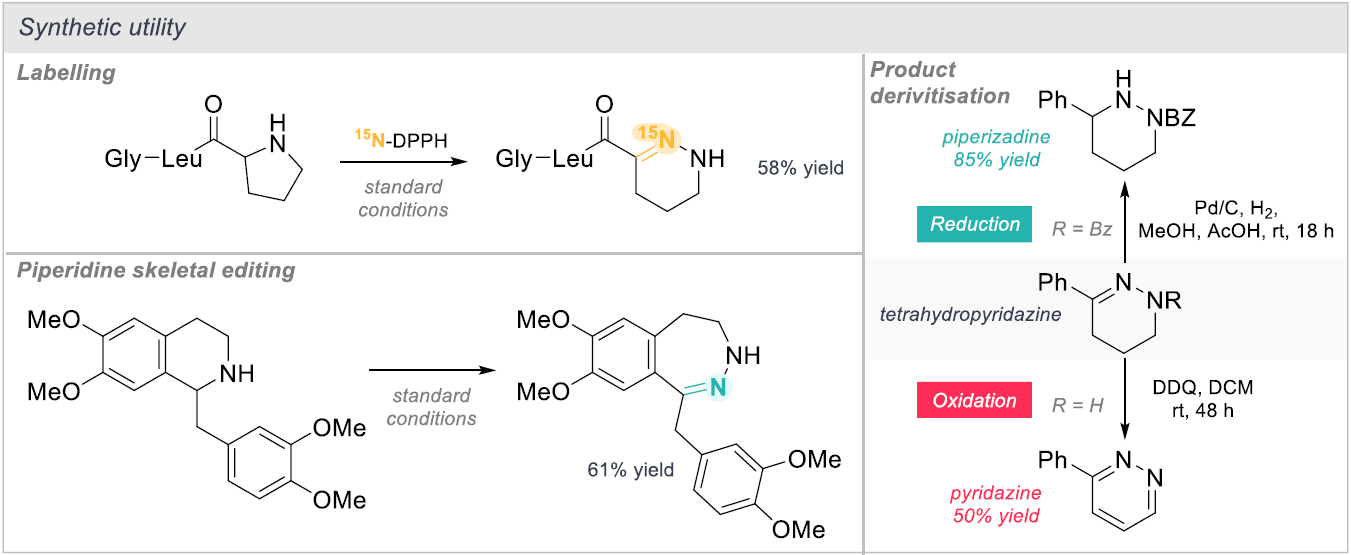
Scheme 3: Skeletal editing and labelling.
In summary, the reported N-atom insertion of pyrrolidine to afford tetrahydrazines presents a powerful method to access scaffolds that were previously inaccessible or highly challenging to construct. This skeletal editing approach offers an exciting opportunity for late-stage functionalisation and diversification of drug-like molecules.
Formal C-H Functionalisation of Quinolines at the C7 Position Through a Traceless Directing Strategy
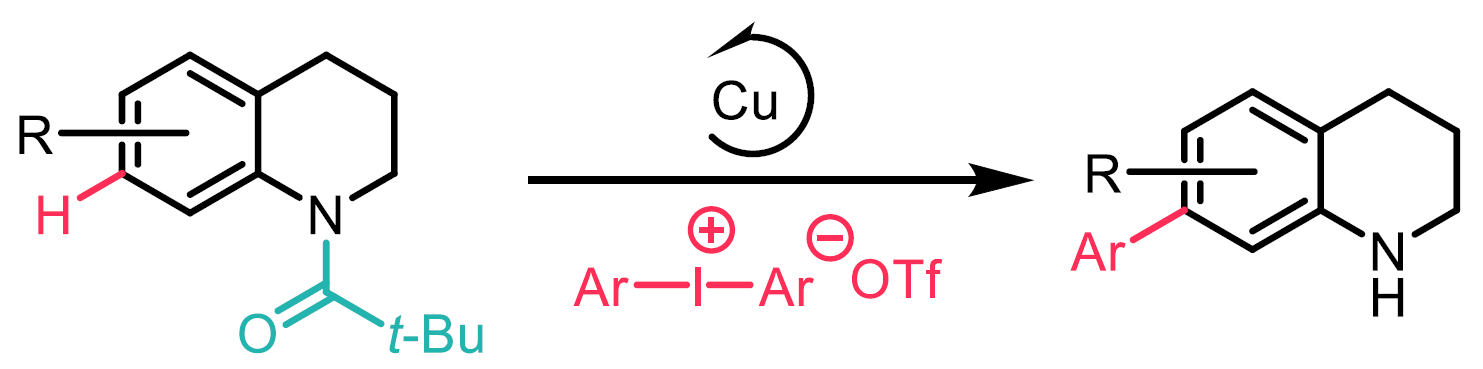
Substituted quinolines are valuable substrates found in ligands, natural products, agrochemicals and pharmaceuticals. Whilst the selective C-H bond functionalisation of the nitrogen-containing cyclic ring of quinolines is well known, the selective C-H functionalisation of the carbocyclic ring is more challenging and predominantly limited to the C8-position via nitrogen to metal coordination. Thus far, selective C-H functionalisation has only been achieved at the C5, C6, and C7 positions using bifunctional templates.1-3 In this work, the authors report a selective copper catalysed formal C-H arylation and alkenylation of quinolines at the C7-position with iodonium triflates using a traceless N-acyl directing group (Scheme 4).
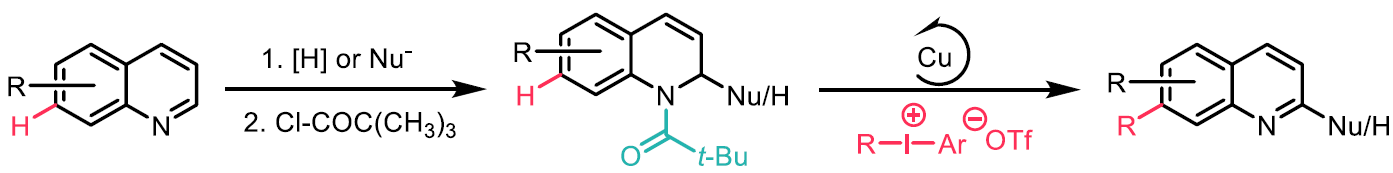
Scheme 4: N-acyl as a traceless directing group for the C-H functionalization of quinolines at C7 position.
It was found that 1,2-hydroquinoline substrates bearing more sterically demanding N-acyl groups performed better, and the best was found to be N-pivaloyl. The choice of diphenyl iodonium salt was found to be essential, significant product formation was only obtained with the triflate salt. The arylation and alkenylation were performed with a range of mesityl-substituted aryl iodonium and alkenyl iodonium triflates, including one containing an estrone moiety. Meta- and para-substitution were well tolerated on the aryl iodonium triflates, but ortho- substitution gave reduced yields due to steric hindrance. Both the arylation and alkenylation were found to tolerate C2, C3, C4, and C5 substitution on the N-acyl 1,2-hydroquinoline, in addition to the more sterically hindered C6 substitution. The selective C7-arylation was successfully extended to 1,2,3,4-tetrahydroquinolines and was found to tolerate C8 substitution. The formation of the C7-free quinoline side-product could be reduced by performing the reaction in a microflow reactor.
Scheme 5: C7-H arylation of 1,2,3,4-tetrahydroquinolines with iodonium triflate using a traceless directing strategy.
Interestingly, it was found that the C7 substituted N-acyl 1,2-dihydroquinolines underwent aromatisation when treated with Cu(OTf)2 and to a lesser extent when treated with Ph2I(OTf)2, whereas the non C7 substituted N-acyl 1,2-dihydroquinolines did not undergo aromatisation at all. Stirring the N-acyl 1,2-dihydroquinolines with HOTf, which would be generated in situ, resulted in formation of the aromatic quinolines in quantitative yield (Scheme 5). Density functional theory (DFT) studies suggest that the N-acyl carbonyl group coordinates to the copper catalyst, placing the aryl group near the C7 position (TS1), which it migrates to. Deprotonation of the C7 position by TfO- gives the arylated product and the N-acyl deprotection then occurs via a six-membered transition state (TS4), in which TfO- attacks the N-acyl group, and is followed by aromatisation (Scheme 6).
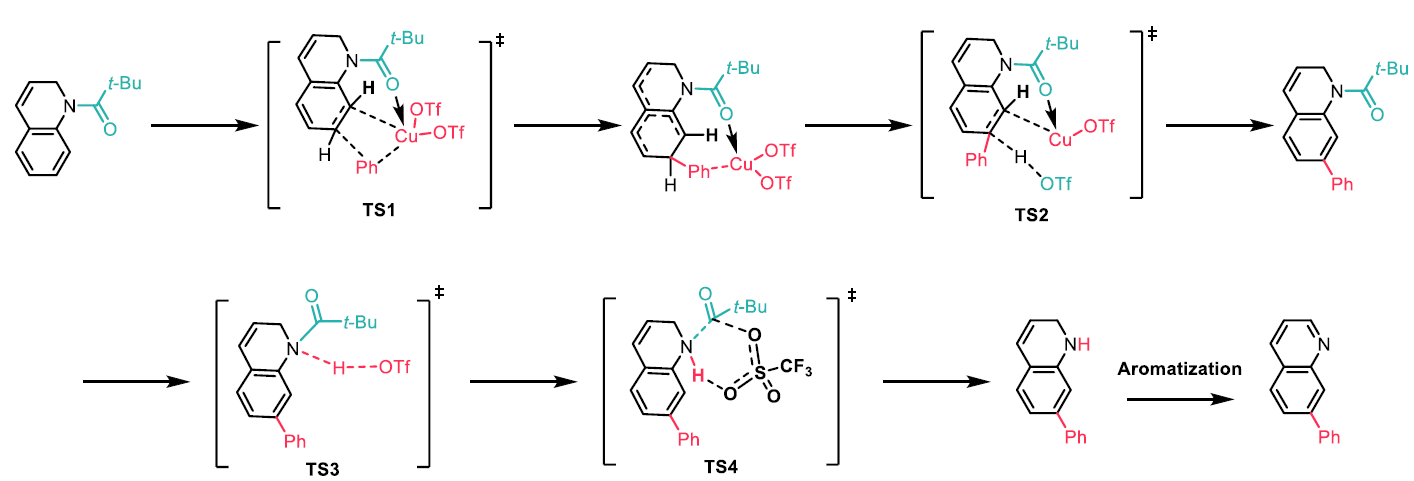
Scheme 6: Proposed mechanism for the C7-H arylation of quinolines based on DFT calculations.
In conclusion, this study presents a significant advancement in the selective C-H functionalisation of quinolines at the elusive C7 position. By employing a traceless N-acyl directing strategy and copper catalysis, the authors have demonstrated efficient arylation and alkenylation using iodonium triflates, with broad substrate scope and functional group tolerance. The mechanistic insights supported by DFT calculations further underscore the role of the N-acyl group in directing reactivity and facilitating deprotection and aromatisation. This methodology opens new avenues for the streamlined synthesis of C7-functionalised quinoline derivatives, with potential applications across medicinal and materials chemistry.
Additional references
- 1 Z. Zhang, K. Tanaka, J.-Q. Yu, Nature 2017, 543, 538–542
- 2 H. Shi, Y. Lu, J. Weng, K. L. Bay, X. Chen, K. Tanaka, P. Verma, K. N. Houk, J.-Q. Yu, Nat. Chem. 2020, 12, 399–404
- 3 Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk, J.-Q. Yu, Nature 2022, 610, 87–93
These two studies exemplify the power of modern synthetic strategies to unlock previously inaccessible chemical space. Skeletal editing through N-atom insertion enables late-stage diversification of saturated heterocycles, while traceless directing group strategies allow for selective C–H functionalisation at challenging positions such as C7 in quinolines. Both approaches significantly expand the toolbox for medicinal chemistry, facilitating rapid scaffold modification and optimisation.
At Domainex, these cutting-edge methodologies will be implemented within our chemistry department to further strengthen our synthetic armory. By integrating these tools into our workflows, we aim to accelerate the design and synthesis of novel, drug-like molecules, ultimately driving discovery and delivering leads.