By Gabriel Negoita-Giras (Associate Principal Scientist, Chemistry)
In the latest edition of our blog series, highlighting exciting new developments in medicinal chemistry, we have focussed on a paper by scientists at Pfizer and Mirati describing the identification of a promising KRASG12D inhibitor; MRTX1133.1 At Domainex we are always looking for great examples of drug design, especially against difficult targets such as RAS. We also have a particular interest in this target as we have previously worked with Professor Terry Rabbits of the University of Oxford on this target (click here to find out more about the collaboration).
Mutant KRAS has long been an attractive target to treat several cancers 2, however, due to a lack of an apparent binding pocket, relatively few inhibitors have been identified to date. Adagrasib (1) was taken as a starting point for this work, which was found to induce and bind to the ‘switch II’ pocket on KRAS. Being a covalent inhibitor, however, Adagrasib is only active in a specific KRAS mutation, which bears a Cys12 residue 3. The team at Pfizer and Mirati set out to design a compound that would selectively inhibit the most common mutation of KRAS, KRASG12D (33%), which would not facilitate a covalent strategy.
The development of MRTX1133 started by removing the Michael acceptor moiety in Adagrasib, thus leaving an unsubstituted piperazine group to pick up an interaction with Asp12. Also, a modified aromatic core was identified, which afforded a 5-10 fold increase in potency and selectivity (2). An X-ray crystal structure confirmed that the binding of 2 with KRASG12D is within a similar ‘switch II’ induced pocket.
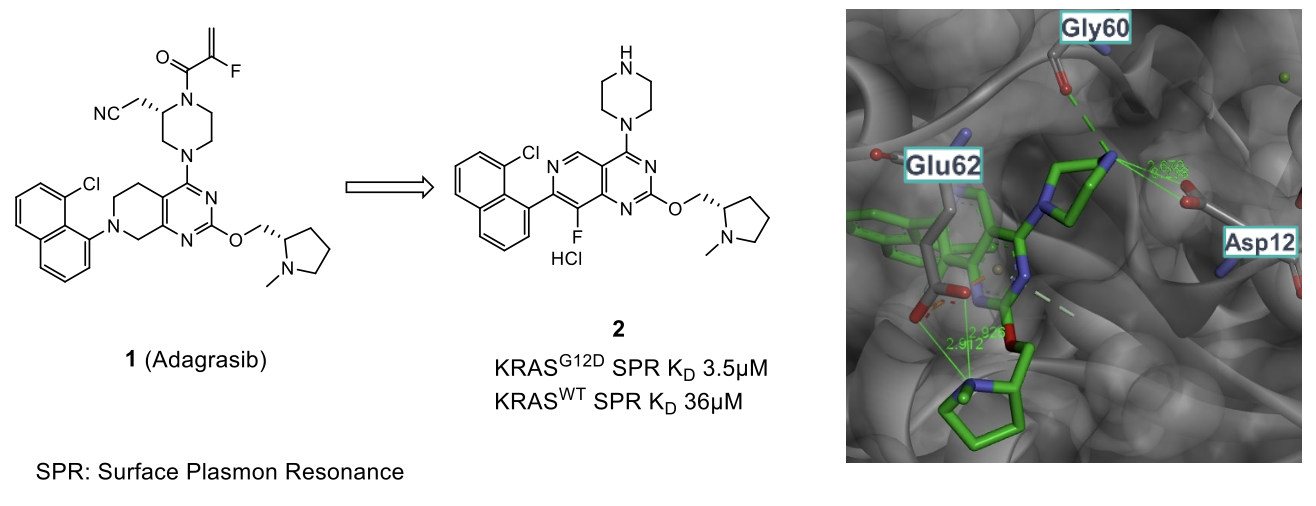
As the covalent binding mode is not feasible, the affinity of the remaining molecule required optimization to make up for the potency lost and maximize the potential of the induced binding site. Three regions coming off the central core were identified and investigated to achieve better binding (R2, R4 and R7).
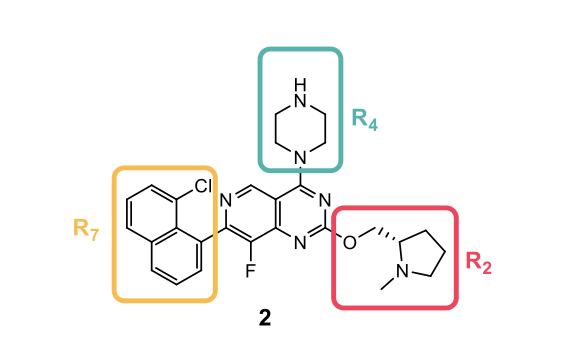
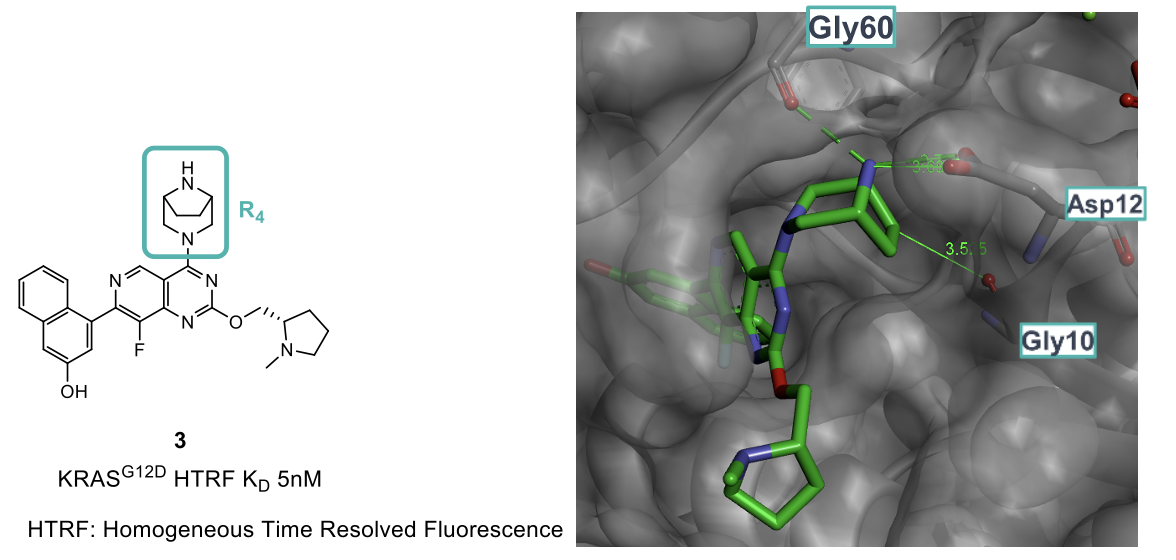
It was found that rigidifying the piperazine R4 group (3) yielded a significant improvement in potency and selectivity by making the predicted interactions with Asp12 and Gly60. Comparison of the crystal structure images exemplifies the gains made as the ‘twist-boat’ piperazine observed for compound 2 is replaced with a much more classical and favored chair conformation. This modification means that the binding mode and most energetically favorable conformation of the small molecule are more aligned, therefore resulting in an improvement in potency. Interestingly, a nonclassical CH-Gly10 interaction was also observed further attributing to the potency uplift.
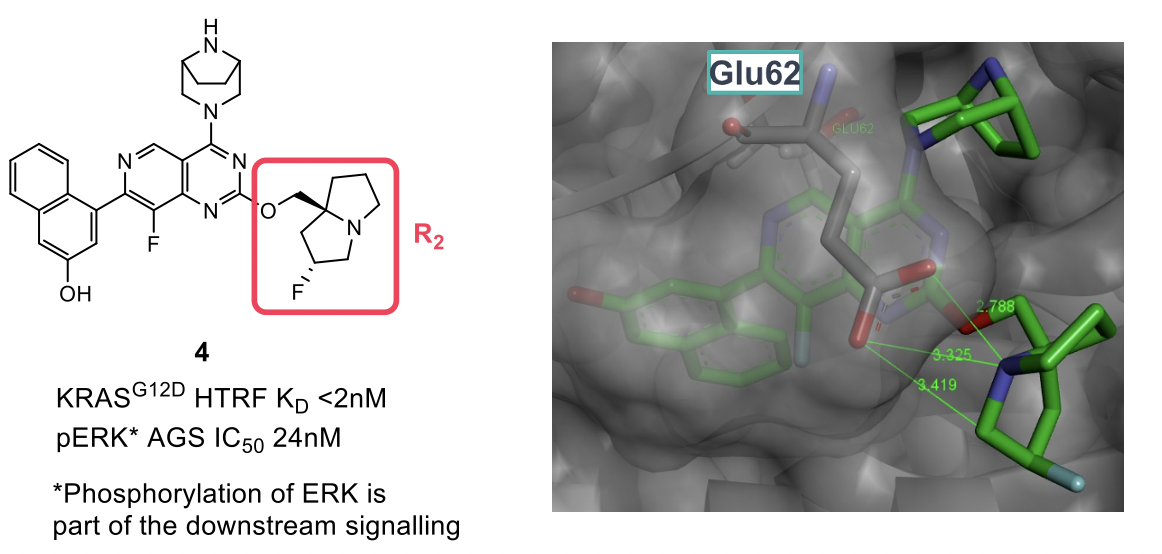
The group then turned to the pyrrolidine R2 basic group coming off the pyrimidine core. Similarly to the previous optimization of the R4 piperazine, it was found that the conformationally strained pyrrolizidine analogue 4 provided superior enzymatic and cellular potencies. Another non-classical CH-Glu62 interaction was observed, and this was thought to be induced by the polarization of the C-H bond by the adjacent fluorine.
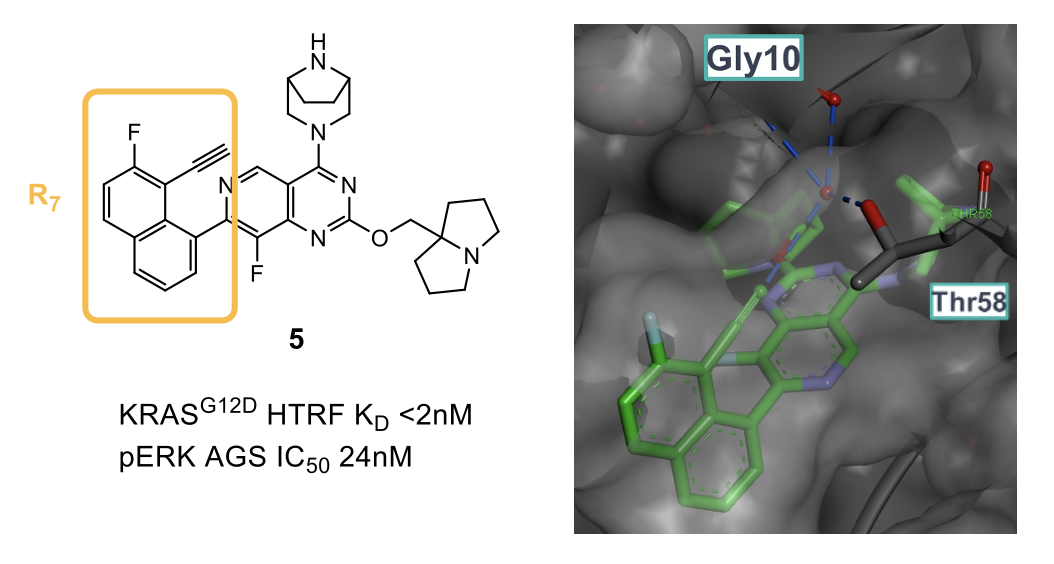
The last group that was optimized was the naphthol R7 group, which occupies a large lipophilic pocket within KRAS. For ease of synthesis, analogues lacking the naphthol group were synthesized, and once the ideal group was identified, the phenol was re-introduced. It was found that the 8 position on the naphthalene ring was optimal for substitution as it allows substituents to better fill the pocket. Furthermore, it was discovered that by introducing an acetylene group in this position (5), a new interaction with a conserved water was picked up, and this in turn stabilized a water mediated bridge between the lipophilic and hydrophilic parts of the pocket (lined by Thr58, Val8 and Gly10).
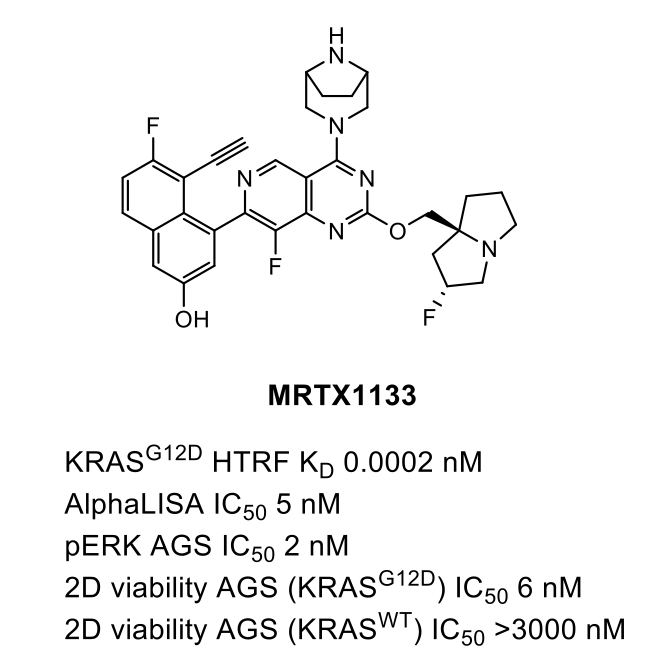
The combination of the three optimized R2, R4 and R7 groups yielded MRTX1133, which optimally filled the ‘switch II’ pocket and benefited from excellent potency and selectivity in enzyme and cellular assays. During our internal discussions at Domainex it was noted that by this point the compound was looking quite lipophilic and indeed most LogP calculators will attribute a value around 5, however, it seems the compound was sufficiently amenable to collect an impressive array of in vivo data.
Following IP administration in xenograft tumor models, MRTX1133 was shown to reduce ERK phosphorylation by 62% (after 1h) and 74% (after 12h) and it proved to be efficacious in an antitumor study by inhibiting growth at low doses (3mg/kg) and inducing tumor regression at higher doses (10mg/kg and 30mg/kg), while no antitumor activity was observed in a non-KRASG12D tumor model.
Through careful and specific optimisation of each area of the molecule, the overall potency was improved 1,000,000-fold from starting molecule 2. In summary, MRTX1133 was identified as a highly potent selective KRASG12D inhibitor with low nanomolar potency across several cellular KRASG12D models, that has the potential to become a first-in-class therapeutic and treat patients with a high unmet need.
Our medicinal chemistry team is always keeping an eye on the latest research to make sure we are up to date with recent developments. The next review will be available soon, but in the meantime, get in touch to find out how we can help solve your drug discovery challenges.
References
- Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor; J. Med. Chem., 2022, 65, 4, 3123–3133
- RAS Proteins and their Regulators in Human Disease, Cell., 2017 Jun 29; 170(1): 17–33.
- Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer; J. Med. Chem., 2020, 63, 13, 6679–6693.